Article Text

Abstract
Background The GNAT1 gene encodes the α subunit of the rod transducin protein, a key element in the rod phototransduction cascade. Variants in GNAT1 have been implicated in stationary night-blindness in the past, but unlike other proteins in the same pathway, it has not previously been implicated in retinitis pigmentosa.
Methods A panel of 182 retinopathy-associated genes was sequenced to locate disease-causing mutations in patients with inherited retinopathies.
Results Sequencing revealed a novel homozygous truncating mutation in the GNAT1 gene in a patient with significant pigmentary disturbance and constriction of visual fields, a presentation consistent with retinitis pigmentosa. This is the first report of a patient homozygous for a complete loss-of-function GNAT1 mutation. The clinical data from this patient provide definitive evidence of retinitis pigmentosa with late onset in addition to the lifelong night-blindness that would be expected from a lack of transducin function.
Conclusion These data suggest that some truncating GNAT1 variants can indeed cause a recessive, mild, late-onset retinal degeneration in human beings rather than just stationary night-blindness as reported previously, with notable similarities to the phenotype of the Gnat1 knockout mouse.
- Retina
- Genetics
- Degeneration
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Transducin is a heterotrimeric G-protein found on the membrane of photoreceptor cells in the retina, and is a key component of the vertebrate phototransduction pathway. On exposure of rod photoreceptor cells to light, the light-sensitive pigment rhodopsin is photoisomerised into its active form. This form activates the G-protein transducin, which in turn stimulates cyclic guanosine monophosphate (cGMP)-phosphodiesterase (PDE). The degradation of cGMP causes cGMP-gated ion channels to close, resulting in hyperpolarisation of the cell membrane and causing the electrical light response.1
Transducin is composed of three polypeptide chains, α, β and γ. The α subunit is the active guanosine diphosphate (GDP)/guanosine diphosphate (GTP)-binding component. On activation by rhodopsin, GTP/GDP exchange occurs and the α subunit dissociates from the βγ subunits; at which point, it becomes capable of binding PDE. The α subunit proteins in rod and cone cells are encoded by the genes GNAT1 and GNAT2, respectively.2
Variants in GNAT1 have been implicated in congenital stationary night-blindness (CSNB) in the past, although never in retinal degenerations. Dryja et al3 reported a missense variant (NM_000172.3:c.113G>A/p.Gly38Asp) causing the dominant ‘Nougaret’ form of CSNB, which was hypothesised to be a result of constitutive activation of transducin. A second example of a pathogenic GNAT1 variant (c.598C>G/p.Gln200Glu) was also reported in a Danish dominant CSNB pedigree.4
GNAT1 variants have also been associated with recessive forms of CSNB. A missense variant (c.386A>G/p.Asp129Gly) was reported to segregate with recessive CSNB in a large, consanguineous Pakistani family.5 The Asp129Gly variant altered a conserved residue of the protein, and was predicted to be deleterious to function, although no functional studies on it were carried out.
We report here a patient with retinal degeneration homozygous for a premature stop codon in the GNAT1 gene (c.904C>T/p.Gln302*). The GNAT1 mutation was identified during the course of a next-generation sequencing (NGS) study covering the Irish patient population with retinal degeneration. This variant causes the elimination of 49 extremely conserved amino acids from the C terminus of the transducin α subunit protein (figure 1), comprising just under one-sixth of the total protein sequence. This is a much more radical alteration to protein structure than that caused by other GNAT1 variants previously associated with CSNB.3–5

Multiple alignment of the amino acids removed by the Q302* variant against the homologous region in other species, showing extremely high levels of sequence conservation, which implies that it is important to protein function.
We have performed an extensive search of the literature and relevant databases, and have not located any other record of a patient with two nonsense GNAT1 alleles. Hence, it is our view that this represents the first report of a homozygous nonsense GNAT1 mutation in human beings, as well as the first observation of retinal degeneration associated with GNAT1 mutations.
This clinical presentation is of particular interest as it strongly resembles the phenotype observed in homozygous knockout (Gnat1−/−) mice. Mice with targeted deletions of the GNAT1 gene have few visible changes in gross morphology, but present with a mild retinal degeneration with age. These mice also show preservation of cone cells, a complete absence of electrical response from rods as well as a slow loss of rod cells with corresponding thinning of the outer nuclear layer,6 features which closely parallel the clinical presentation in the GNAT1 Gln302* patient.
Materials and methods
Patient identification and recruitment
The proband was recruited prospectively at the Research Foundation, The Royal Victoria Eye and Ear Hospital, Dublin, as part of the Target 5000 study, an ongoing project to identify the molecular basis for inherited retinopathies in Irish patients via NGS.
Clinical assessment
Best-corrected visual acuity was assessed using revised 2000 Early Treatment Diabetic Retinopathy Study charts (Precision Vision, La Salle, Illinois, USA). Colour vision was examined using the Lanthony desaturated panel D-157 under standardised lighting conditions. Goldmann perimetry was used to assess the peripheral visual fields to the IV4e, I4e and 04e targets. Following slit lamp biomicroscopy of the anterior segment of the eye and Goldmann applanation tonometry, the patient's pupils were dilated using tropicamide 1% and phenylephrine 2.5%. The subject was dark-adapted for 30 min, and the dark-adapted threshold was then measured using a Goldmann–Weekers dark adaptometer. A full-field electroretinogram (ERG) was performed according to International Society for Clinical Electrophysiology of Vision (ISCEV) standards8 using a Roland Consult RETI-port RETIscan. Fundus photography was performed using a Topcon CRC50DX. Spectral domain optical coherence tomography was performed using a Cirrus HD-optical coherence tomography (OCT) (Carl Zeiss Meditec, Germany).
DNA isolation and sequencing
Following informed consent, blood samples were collected from patients after clinical assessment. DNA was isolated from 2 mL of patient blood using the QIAamp DNA Blood Midi kit (QIAGEN, Valencia, California, USA), according to manufacturer's recommendations.
DNA was fragmented for sequencing by ultrasonication in a Diagenode Bioruptor (Diagenode, Belgium) for 35 cycles, each consisting 30 s of low-intensity sonication followed by a 30 s pause. Water was replaced and crushed ice added regularly to maintain a sonication temperature of <4°C.
Sequencing libraries were generated and target capture was performed using the Agilent SureSelect XT2 kit (Agilent Technologies, Santa Clara, California, USA), according to manufacturer's recommendations. Exons and untranslated regions (UTRs) for all genes previously implicated in retinal degeneration as of early 2013, as listed by Retnet, were included as capture targets: a total of 3592 capture regions across 182 genes, spanning a total sequence size of 1.25 Mb, of which, 497 kb were coding exons, and the remainder UTRs (see online supplementary table S1).
Supplementary table
Captured patient DNA was multiplexed into a 96-sample pool, and sent for sequencing. Sequencing was performed off-site by BGI Tech using an Illumina HiSeq 2000 (Illumina, San Diego, California, USA). Confirmatory single-read sequencing was also performed to verify the presence of candidate mutations.
Data analysis
Sequence data were demultiplexed and mapped to the human genome (hg19) using BWA V.0.7.4.9 Duplicate reads were flagged using Picard V.1.106,10 and downstream analysis and variant calling were performed using the GATK V.2.811 according to the protocol specified in the GATK Best Practices Workflow.12
The list of identified variants was annotated with snpEFF13 and dbNSFP.14 Synonymous and common variants were filtered out, and the remaining list of rare variants with the potential to affect protein sequence was output for manual curation.
Results
Clinical findings
The proband presented at 80 years of age with a 6-month history of difficulties with steps. He commented on a lifelong history of night-blindness, which had remained unchanged throughout his life. There was no family history of night-blindness or day vision problems affecting either his parents, three siblings, ranging in age from 73 to 75 years or his three children aged 38–51 years. There was no history of parental consanguinity. His general health was good. He suffered for 10 years prior to presentation with non-insulin dependent diabetes mellitus, which was well controlled.
On examination, the proband's unaided visual acuity was 6/7.5 in the right eye, which improved to 6/6 with +0.5 ds/+0.5 cx at 165°, and 6/7.5 in the left, which improved to 6/6 with 0.0/+0.5 cx at 165°. Slit lamp examination revealed healthy corneas, normal anterior segments and clear lenses apart from some peripheral cortical opacities. His vitreous was normal apart from the presence of a grade 1 cellular infiltrate. Intraocular pressures were normal at 14 mm Hg in the right eye and 14 mm Hg in the left.
The proband's dark-adapted threshold to an 11° target was assessed at 15° from fixation in each eye using a Goldmann–Weekers dark adaptometer. Thresholds were significantly elevated at 4.3 log units in the right eye and 4.1 log units in the left, the upper limit of normal for our laboratory being 2.0 log units.
A full-field ERG, performed to ISCEV standard, revealed no convincing rod-isolated responses. Convincing ERG responses were recorded to the maximal intensity flash recorded in the dark-adapted state (figure 2). These responses are usually mediated by both the rods and cones. Clear cone-mediated responses were recorded to both single flash and 30 Hz flicker stimulation, in each case, against a rod-suppressing background illumination. These responses were significantly delayed and attenuated in amplitude (figure 2). Spectral-domain OCT showed an epiretinal membrane bilaterally with preservation of the foveal contour (figure 3).

Full-field electroretinograms in the proband showing a lack of rod phototransduction. Tracings ‘a’ and ‘b’ show non-recordable dark-adapted rod-isolated responses in the right and left eyes, respectively. Tracings ‘c’ and ‘d’ show significantly attenuated dark-adapted responses to the maximal intensity flash. Ordinarily, these responses reflect rod and cone activities. Owing to the absence of rod-isolated inputs, these responses are most probably cone-driven in our proband. Tracings ‘e’ and ‘f’ show significantly attenuated light-adapted cone-isolated responses.

Above: Fundus photographs of the proband’s right and left eyes, showing optic disc pallor, arteriolar attenuation, normal maculae and scattered bone spicule pigment deposits. Below: OCT images of the proband’s right and left eyes, showing normal foveal contour with some thickening and epiretinal membrane.
Fundus examination showed slight disc pallor and arteriolar attenuation in each eye. Both maculae appeared healthy. Marked peripheral pigmentary deposits, some of which were bone spicule in appearance and some with a more clumpy appearance, were evident in the retinal peripheries (figure 3). There was no evidence of diabetic retinopathy in either eye.
One of the proband's children was also assessed. As the proband was homozygous for the GNAT1 variant, his daughter was an obligate carrier, and presence of the heterozygous variant was confirmed by Sanger sequencing.
The proband's daughter was 56 years of age at the time of assessment. She had no complaints of night-vision or day-vision difficulties. Her dark-adapted thresholds were normal at 1.4 log units in each eye. Full-field ERGs showed normal rod-isolated and cone-isolated responses in both eyes. Detailed funduscopic examination was entirely normal in each eye, and visual fields did not show the constriction evident in the proband (figure 4). The OCT of the proband's daughter was normal.

Visual fields for both eyes of the proband (below) and unaffected offspring (above). The proband shows clear constriction of visual fields consistent with a diagnosis of retinitis pigmentosa.
Genetic studies
The GNAT1 Gln302* variant was first identified in the proband during analysis of NGS data. Sequencing quality for the proband's sample was overall very good: 98.3% of bases in coding regions and 94.3% of bases overall were considered ‘callable’ with a coverage of at least 10×, and 57.3% of reads were ‘on target’ (overlapping a capture region). Confirmatory Sanger sequencing confirmed the presence of the homozygous variant. This variant eliminates 49 amino acids from the C terminus of the GNAT1 protein, approximately one-sixth of the total protein length (figure 1).
The variant was not observed, even heterozygously, in 190 other patients involved in this study, 4 of whom had been diagnosed with CSNB. No other GNAT1 mutations were observed in any patients involved in this study. The variant was not observed in the 1000 Genomes project,15 but was observed at an extremely low allele frequency of 5/66 210 in European populations by the Exome Aggregation Consortium,16 with no homozygous cases and no observations of the allele in other ethnic groups. A literature search of PubMed and Human Gene Mutation Database17 did not reveal any previously reported cases of any other homozygous truncating GNAT1 variant. Sequencing of the exons of 181 other retinopathy-associated genes in this patient did not reveal any other variants that were considered to be plausibly responsible for the observed phenotype. Sequencing of the unaffected daughter detected the variant to be present heterozygously, but without any disease phenotype, suggesting a recessive mode of disease inheritance.
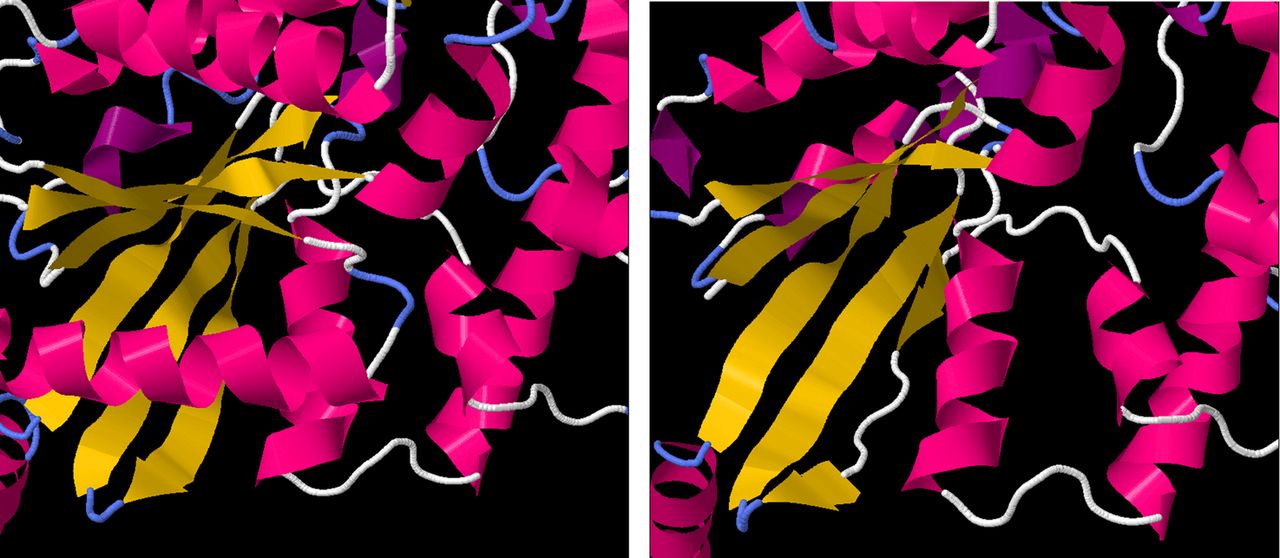
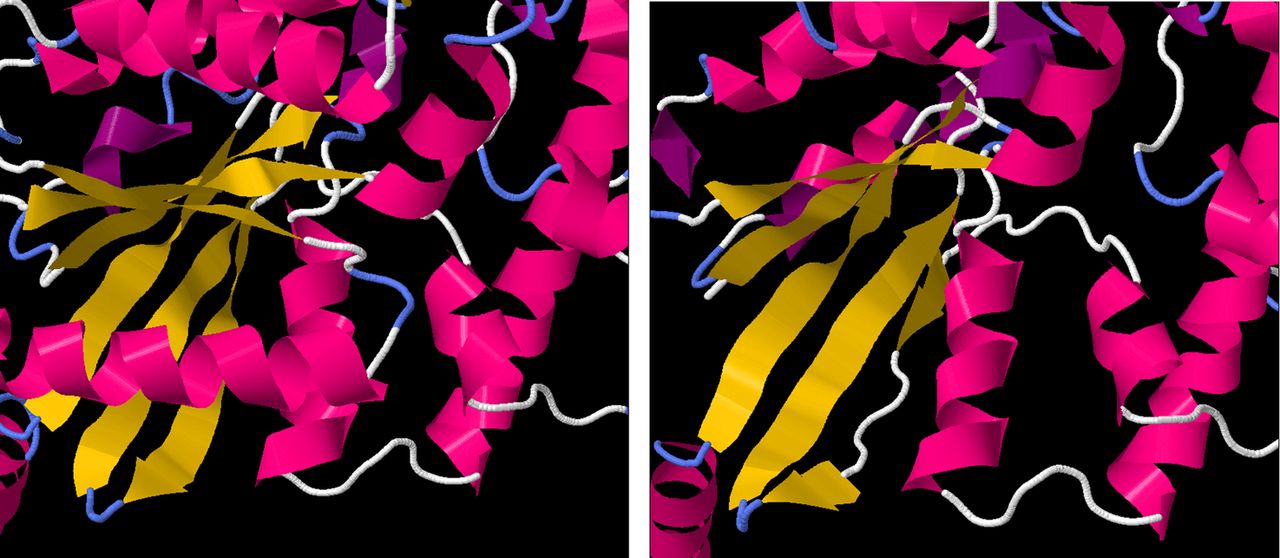
Several of the deleted residues are crucial to α-transducin protein function. Bonds between Thr 323 and Lys 266 are involved in forming the guanine-binding pocket.18 Residues 311–329 have been shown to bind photoactivated rhodopsin,18 while residues 306–311 and 314 are involved in PDE activation.19 Simulated folding of the truncated peptide performed using the I-TASSER software suite20 suggested that the six-stranded β-sheet GTPase domain would be destabilised by the deletion, with only four of the strands forming correctly and a fifth forming incompletely, while the sixth not forming at all (figure 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Left: the protein structure of GNAT1, derived from X-ray crystallographic measurements. The α-helical regions are coloured in pink, while β sheets are coloured in yellow. The six-stranded β-sheet domain, central to GTPase function, is fully formed. Right: the predicted structure of the truncated GNAT1 protein, showing the malformations in the fifth and sixth strands of the β-sheet domain.
As GTPase activity is crucial to the function of all G-proteins, the GNAT1 Gln302* variant is, therefore, predicted to very severely affect protein function, impacting on the protein's interactions with both upstream and downstream members of the signalling pathway.
There is a substantial difference in phenotype between the proband and other reported patients with CSNB caused by GNAT1 variants. In addition to the expected lifelong night-blindness, the GNAT1 Gln302* patient presented with a late-onset retinitis pigmentosa-like degeneration of the retina, including bone spicule deposits. In contrast, Naeem et al5 explicitly stated that they did not observe any such retinitis pigmentosa-like features on fundus examination of patients who were homozygous for the Asp129Gly variant. While retinal degeneration is clearly a feature of the GNAT1 Gln302* disease, it is slow; at age 80, the patient still has good central and colour vision.
Discussion
There are interesting parallels between the observed clinical presentation of the homozygous GNAT1 Gln302* mutation and the phenotype of the Gnat1 knockout mouse. No convincing rod-isolated ERG responses were recorded from the proband to the −25 dB flash stimulus, providing an explanation for the subject's history of night-blindness and consistent with the elevated dark-adapted thresholds. Similarly, Calvert et al6 could not record any rod-isolated ERG responses in mice with a targeted deletion of the GNAT1 gene. Retinal excitation by flash stimuli, sufficiently bright to elicit mixed rod and cone responses from the healthy retina, only elicited responses compatible with cone stimulation alone in both the proband and in Gnat1−/− mice.
The pattern of the dark-adapted electroretinographic responses in the GNAT1 Gln302* patient differs significantly from many patients with CSNB, where there is an ‘electronegative’ conformation to the dark-adapted mixed rod/cone response to the maximal intensity flash stimulus, but are similar to those reported in autosomal recessive5 and autosomal dominant21 forms of CSNB caused by GNAT1 mutations. Notably, however, other GNAT1 CSNB kindreds did not show the clear pigmentary disturbances observed in the Gln302* patient.
Cone-isolated ERG responses in the Gnat1−/− mice, when assessed at 8–10 weeks, showed no appreciable difference compared with age-matched wild-type controls.6 The cone-isolated responses in the GNAT1 Gln302* patient, at 80 years of age, showed delayed and reduced amplitude responses (figure 2), indicating significantly compromised cone function. Calvert et al6 did not present data on cone ERG responses in Gnat1−/− mice of an age equivalent to that of the proband in the current study.
It is particularly noteworthy that mice homozygous for the GNAT1 deletion showed a subtle thinning of the retina over time.6 By 13 weeks of age, rod-outer-segment length shortens, and the thickness of the outer nuclear layer decreases by about one row of nuclei, indicating a loss of approximately 10% of rod cells, followed by a later mild loss of thickness in the inner nuclear layer.6 The rate of decline in the thickness of the outer nuclear layer slowed substantially after the first 13 weeks.
Despite this decline in retinal thickness, the authors predicted that deletion of GNAT1 in human beings would cause a recessive stationary night-blindness rather than retinitis pigmentosa. The proband, however, clearly shows cone photoreceptor dysfunction on electroretinographic examination and visible funduscopic signs indicative of a mild and localised, but definite, retinitis pigmentosa.
Given the above observations, we conclude that the novel Gln302* mutation in the GNAT1 gene may indeed cause a slow and late onset form of retinitis pigmentosa in human beings, and hypothesise that slow loss of rod cells with age, similar to that observed in the Gnat1−/− mouse, may be responsible for this.
In all previously reported cases of GNAT1-mediated CSNB, the causative mutations have been single amino acid substitutions, rather than truncating or frameshift mutations. This is the first case of a homozygous variant introducing a premature stop codon into GNAT1, eliminating crucial domains from the GNAT1 protein. In addition, this study represents the first evidence that GNAT1 mutations can cause retinal degeneration in human patients. Until now, transducin was unusual in the rod phototransduction pathway in that it had not been associated with retinal degeneration; variants in both rhodopsin, the immediately upstream component of the pathway and cGMP-PDE, the immediately downstream component, have been implicated in retinitis pigmentosa.22–24
It is important to note that the GNAT1 Gln302* patient developed retinitis pigmentosa at a late age, older than many of the patients assessed in prior studies of CSNB caused by GNAT1 variants.3–5 We, therefore, cannot exclude the possibility that the observed phenotype is not confined to this or other truncating variants, and may occur in other patients with GNAT1-mediated CSNB as they advance in age. We recommend that clinicians monitor older patients with GNAT1-mediated CSNB for evidence of degeneration. We also recommend that GNAT1 should be considered as a candidate for sequencing in patients with recessive or simplex lifelong night-blindness and late-onset retinal degeneration.
Acknowledgments
The authors would like to acknowledge grant awards from Fighting Blindness Ireland, The Health Research Board, The Medical Research Charities Group and Science Foundation Ireland.
References
Footnotes
Contributors MC performed sample preparation, design of target panels, sequencing and bioinformatics analysis, as well as primary authorship of the manuscript. ED and PFK performed clinical assessments, sample collection, analysis of sequencing results and location of additional pedigree members, as well as manuscript preparation. PFK was also involved in the initial study design. AP was involved in data analysis and preparation of the manuscript. PH and GJF were involved in study design and manuscript preparation. GJF was the overall supervisor of the research group.
Funding Fighting Blindness Ireland, Medical Research Charities Group, Science Foundation Ireland, Health Research Board.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethics and Medical Research Committee, Royal Victoria Eye and Ear Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- At a glance