Article Text

Abstract
The landmark publications that gave such impetus to our understanding of proliferative diabetic retinopathy are reviewed in the light of more recent reports. Briefly, confluence of small areas of capillary closure in the midperipheral and peripheral retina results in arteriovenous shunting and abnormal oxygen partial pressure gradients. These gradients embrace a chronic ischaemic penumbra that stimulates neuroglial secretion of angiogenic growth factors and upregulation of their receptors in the retinal venous endothelium and adventitia. The blood shunting produces biomechanical stresses within the veins and induces microvascular intussusception near arteriovenous crossings, giving way to neovascular outgrowths and/or segmental venous lesions (such as omega loops and coils) that penetrate the inner limiting lamina. The lamellar collagenous matrix of the vitreous cortex is then exploited for integrin-dependent rete expansion along chemotactic gradients. During posterior vitreous detachment, haemorrhaging takes place from the arterialised veins as venous neovascular peduncles are avulsed.
- CRA, central retinal artery
- CRV, central retinal vein
- ECM, extracellular matrix
- ERM, epiretinal membrane
- FFA, fundus fluorescein angiography
- GMP, grey matter penumbra
- ILL, inner limiting lamina
- MVI, microvascular intussusception
- ODNV, optic disc new vessel
- PDR, proliferative diabetic retinopathy
- PHM, posterior hyaloid membrane
- PlGF, placental growth factor
- PRNV, preretinal new vessel
- RAV, radiating anastomosing vessel
- VEGF, vascular endothelial growth factor
- VEGFR, vascular endothelial growth factor receptor
- VNP, venous neovascular peduncle
Statistics from Altmetric.com
- CRA, central retinal artery
- CRV, central retinal vein
- ECM, extracellular matrix
- ERM, epiretinal membrane
- FFA, fundus fluorescein angiography
- GMP, grey matter penumbra
- ILL, inner limiting lamina
- MVI, microvascular intussusception
- ODNV, optic disc new vessel
- PDR, proliferative diabetic retinopathy
- PHM, posterior hyaloid membrane
- PlGF, placental growth factor
- PRNV, preretinal new vessel
- RAV, radiating anastomosing vessel
- VEGF, vascular endothelial growth factor
- VEGFR, vascular endothelial growth factor receptor
- VNP, venous neovascular peduncle
The growth of preretinal new vessels (PRNVs) and optic disc new vessels (ODNVs) in proliferative diabetic retinopathy (PDR) represents an aberrant angiogenic response to ischaemia of the inner retina (fig 1).1 This essay revisits the pivotal publications that advanced or substantiated the key hypotheses surrounding PDR.
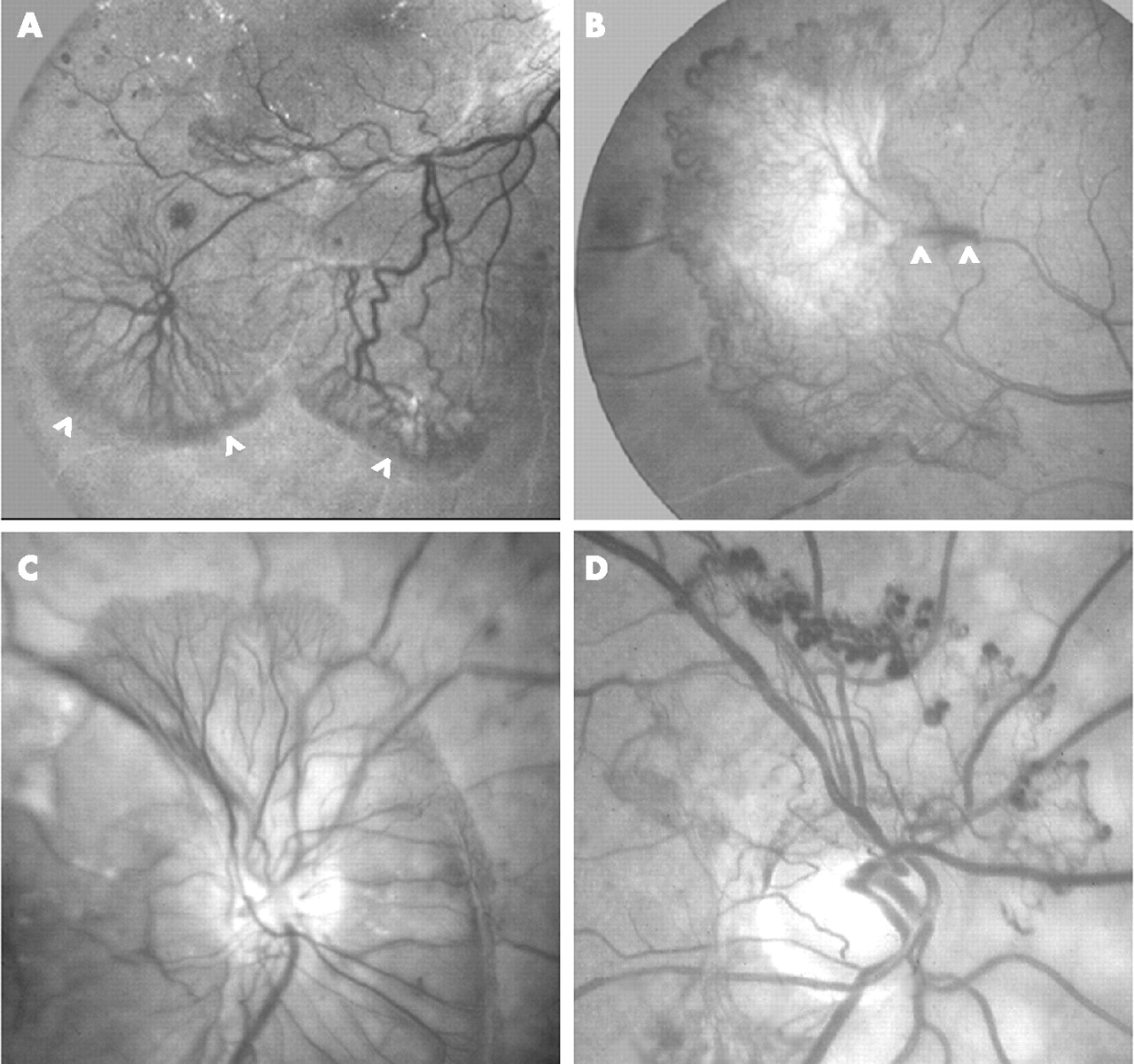
Preretinal and prepapillary new vessels. (A) Two rete expansions directed inferotemporally towards the ischaemic periphery; truncated cartwheel configuration of preretinal new vessels and brushes of delicate terminal loops (arrowheads). (B) Fibrovascular epiretinal membrane directed temporally towards the ischaemic periphery; marginal vessels; elongated peduncle (between arrowheads) arising from arteriovenous crossing. (C) Multiple feeder vessels from the optic disc serve rete expansion within preretinal vitreous cortex; brushes of terminal loops. (D) Diaphanous rete of peripapillary optic disc new vessels with aneurysmal dilatations at the limit of rete expansion.
VENOUS LOOPS, RETE MIRABILE AND MICROVASCULAR INTUSSUSCEPTION
In 1946, Arthur Ballantyne2 provided one of the first clinicopathological descriptions of PDR. He observed endothelial cell proliferation throughout the retinal venous system together with hyaline thickening (phlebosclerosis) of the walls of those beaded veins from which new blood vessels and segmental venous abnormalities originated. Focal erosions of the internal limiting membrane then permitted the emergence of discrete neovascular projections into the vitreous cavity, whereas much wider dehiscences accommodated venous coils, knots and omega loops, as shown by serial retinal sectioning.2,3 These segmental lesions were subsequently dubbed “benign forms of retinitis proliferans”, and emphasise the scope of Ballentyne’s vasoproliferative concept.4
Although early PRNVs appeared “naked” on fundoscopy, histopathological examination of these outgrowths disclosed a prominent interstitial component which Ballantyne attributed to “metaplasia of the tissue of the vein wall”. This component was evident well ahead of the wave of fibroblast proliferation and collagen synthesis marking the second, fibrotic clinical phase of PDR.2,5–8 Meanwhile, he applied the term rete mirabile to the network that is formed when a venous outgrowth divides into a substantial number of branches that ultimately reunite into a single stem. These neovascular expansions, comprising vessels of capillary structure but of far greater diameter, frequently terminated in a brush-like arrangement of loops.
The fact that PRNVs sometimes arise from segmental venous lesions, and the capability of both pathologies to dissolve the inner limiting lamina (ILL), suggest that similar pathogenetic mechanisms are operating.3–5,9 Each is compatible with intussusception, a recently expounded concept of microvascular growth, branching and remodelling based on internal division of the parent vessel (“growth from within”). Thus, rearrangement of endothelial cells and intrusion of a pillar of interstitial tissue comprising fibroblasts, myofibroblasts and pericytes leads to partitioning of a vein’s lumen. Alternatively, fusion of luminal evaginations on either side of a partial tissue septum establishes a patent loop within the vein wall (fig 2 A,B).10–13 Elongation of such loops and generation of new vascular elements extend the process, while collagen deposition provides mechanical stability. All channels remain perfused unless thrombosed or closed by cellular invasion during remodelling or regression.

Microvascular intussusception. (A–D) Diagrams after Patan et al;13 arrows indicate direction of blood flow. Lumen (grey) of the parent vein evaginates (A) around a partial interstitial septum (black) forming a perfused loop in the vein wall (B). Lateral intussusception creates a vascular network or rete mirabile connected to the parent vein by afferent and efferent channels within the venous neovascular peduncle (C). Parallel or lengthwise intussusception elongates the loop and creates venous reduplication (D). (C′) Small rete mirabile. (D′) Inflated omega (Ω) loop spanning an arteriole; arteriovenous anastomosis (between white arrowheads) and putative venous partitioning (black arrowhead).
A novel hypothesis encompassing Ballantyne’s observations is therefore proposed. Predominantly lengthwise progression of partitioning along the course of the parent vein gives rise to loops, coils and parallel channels (venous reduplication), whereas arborisation directed laterally away from the trunk vessel results in formation of a rete mirabile (fig 2). Lateral microvascular intussusception (MVI) is more energy efficient than “sprouting”, the classical mode of angiogenesis, since this requires endothelial cells to proliferate and form blind-ending tubes that must stabilise and fuse with each other to allow rete perfusion.14,15 A blood-filled lumen extending to the distal limit of the neovascular networks, and thus implicating MVI, has been demonstrated in serial reconstructions of excised diabetic preretinal tissue.16
MVI readily explains how venous neovascular peduncles (VNPs) are generated. Each incorporates an afferent and efferent stem connecting the rete mirabile to the host vein (fig 2C). The minimal hydrostatic pressure gradient thus available to drive perfusion through the dilated veno-venous plexus accounts for the slow flow of dye observed during fundus fluorescein angiography (FFA),8,17 a property overshadowed by the profuse dye leakage. Where adjacent networks anastomose, however, vascular remodelling can be envisaged to redirect and accelerate the flow and to render any one (or even both) of the connecting channels in any VNP redundant.
HYPOXIC UPREGULATION OF ANGIOGENIC GROWTH FACTORS AND THEIR RECEPTORS
In 1948, Isaac Michaelson18 reported his Indian ink injection studies on the fetal retinal vasculature. He observed a periarterial capillary-free zone as vascular networks emerged from the venous side of the developing circulation and felt this implied a gradient of progressive retinal hypoxia with increasing distance from the artery. This led to his presenting his celebrated hypothesis implicating a hypoxia-inducible “environmental factor” that controls not only embryonic vessel formation (either de novo from precursor cells, “vasculogenesis”, or from pre-existing vessels, “angiogenesis”) but also pathological neovascularisation of the mature retina.
Michaelson suggested that, in PDR, the soluble mediator is secreted by extravascular retinal components challenged by hypoxia from a circulatory disturbance yet to be discovered. Moreover, in the published discussion of his presentation, he introduced the concept of “remote” neovascularisation by postulating that diffusion of the same molecule throughout the vitreous cavity, overspilling into the posterior and anterior aqueous chambers, stimulates rubeosis iridis. Iris neovascularisation and formation of ODNVs and PRNVs are now believed to involve recruitment of circulating endothelial progenitor cells (“adult vasculogenesis”) as well as angiogenesis.19
Extracts of vitreous gel removed from eyes with PDR induce directional vessel growth in vivo in the chick chorioallantoic membrane20 and are a ready source of candidates for Michaelson’s “factor x”. Angiogenic molecules thus far isolated include insulin-like growth factor 1, endothelial cell-stimulating angiogenesis factor, fibroblast growth factor, vascular endothelial growth factor (VEGF), placental growth factor (PlGF), hepatocyte growth factor, erythropoietin and angiopoietin.21–29 The vitreous concentration of any such factor at any one time is the resultant of its retinal release set against its diffusion utilisation, enzymatic destruction and sequestration on cell membranes or extracellular matrix (ECM) components. The endocrine “growth hormone/insulin-like growth factor 1 axis” is also crucial to intraocular neovascularisation, witness the effect of pituitary ablation, while vitreous levels of connective tissue growth factor reflect the extent of associated fibrosis.30,31
The primacy of the role of VEGF in PDR is suggested, first, by its generic properties in relation to Michaelson’s “chemical mediator hypothesis”. VEGF receptors (VEGFRs) are hyperexpressed on retinal vascular endothelial cells, and synthesis of VEGF protein (and that of its receptors) is modulated by the local oxygen tension.32–34 The efficient cellular secretion of all its isoforms and free diffusibility of smaller isoforms further combine to make VEGF an ideal candidate molecule. Secondly, indirect evidence of a substantial role for VEGF in PDR includes its involvement in angiogenesis in hypoxic tissues and in tumours generally, whether through intussusception or sprouting or both.13,14 Moreover, intravitreal injection of VEGF induces rubeosis iridis and retinal MVI that penetrates the ILL, while other types of experimental ocular angiogenesis can be inhibited by targeting action of VEGF receptor.35,36
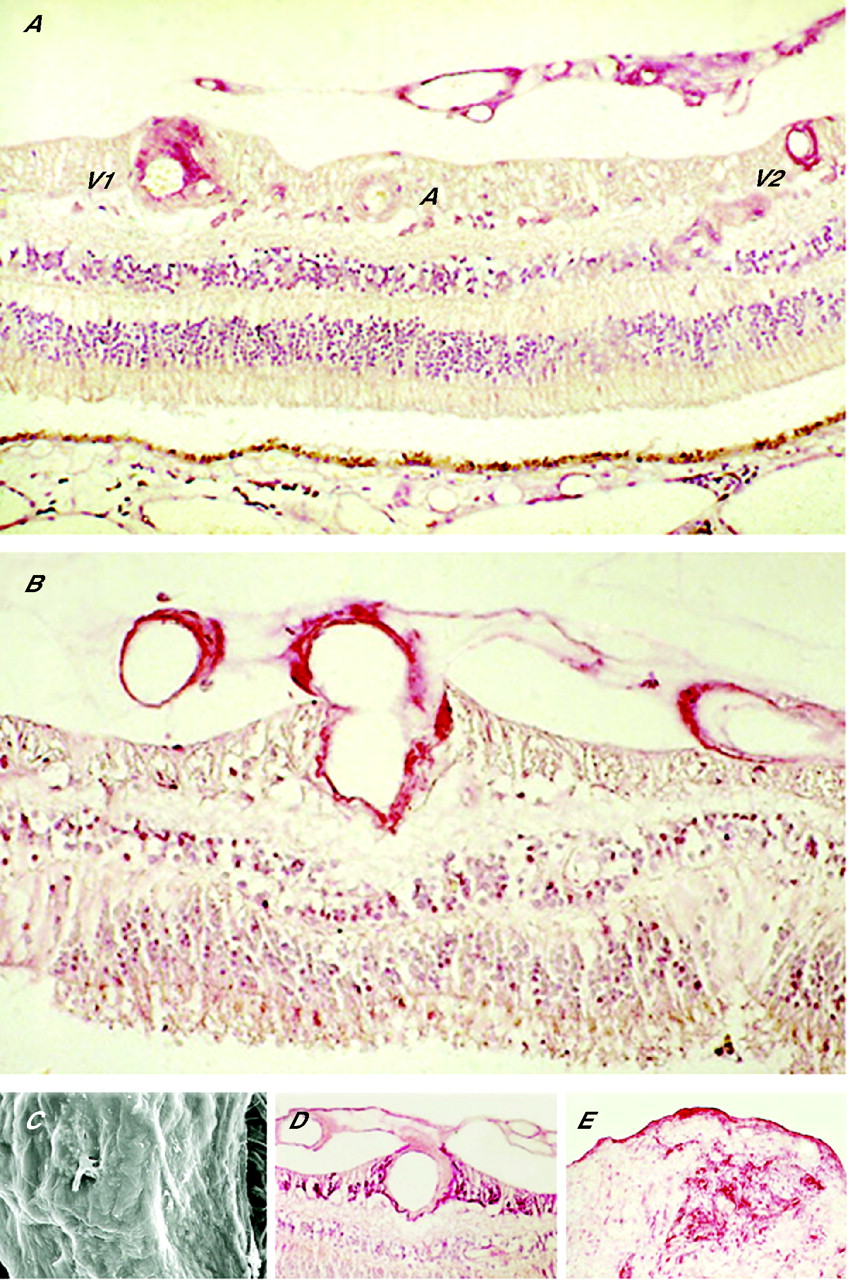
Human histopathological examination provides a third and direct source of evidence that VEGF is a vital mediator in PDR (fig 3). Retinal VEGF immunostaining increases with increasing severity of retinopathy,37,38 but this is not a sufficient indication of the protein’s role. Increased bioactivity depends on the ligand’s access to, and the density and avidity of, the relevant receptors on its target cells. A spatial and temporal correlation can be shown between the retinal expression of VEGF and its main receptor, VEGFR2, as the retinopathy progresses.38,39 Eventually, the VEGF/VEGFR2 system is massively expressed within and immediately surrounding those veins, and giving rise to the new vessels as well as, in continuity with the preretinal neovasculature itself. Similar spatial and temporal coupling applies to the PlGF/VEGFR1 system that has been implicated in adult vasculogenesis.26,39,40 Molecular signals from other participating systems no doubt interact to stimulate, modulate and terminate the various stages of MVI.

Immunochemistry and ultrastructure in proliferative diabetic retinopathy (PDR). (A) Retina with PDR stained for vascular endothelial growth factor protein; intense staining of preretinal new vessels and retinal veins (V1 and V2) in cross-section, but the retinal artery (A) is unstained. Loss of ganglion cell layer, inner nuclear layer and part of the outer nuclear layer midway between A and V2 (representing “core of infarction”). (B) Retina with PDR stained for vascular endothelial growth factor receptor 1; intense staining of endothelium and adventitia of intraretinal vein and preretinal neovasculature in continuity. (C) Scanning electron microscopy of an excised segment of fibrovascular epiretinal membrane (ERM); smooth undersurface interrupted by vessel. (D,E) Glial fibrillary acidic protein staining of the retina with PDR (D) and excised fibrovascular ERM (E): glial component of peduncle, stroma and inner (vitreous) surface lamina (figs 3A,B and D from Boulton et al38 and Smith et al39).
Once new vessels penetrate the ILL, their further ramifications are orientated parallel to the retinal surface, and especially towards the hypoxic retinal territories (fig 1A,B). Such “directionality” is probably determined by the distribution of VEGF in the ECM, as either variable concentrations of soluble isoforms in the gel or deposits of large heparin-binding isoforms on vitreous fibrils. Specialised migrating endothelial cells or “tip cells” in the vanguard of the neovascular plexus have been implicated in rete expansion along these putative chemotactic gradients.41 A truncated “cartwheel” centred on a VNP is the characteristic angiogenetic pattern in PDR, while the delicate loops at the distal limit of vasoproliferation are sometimes augmented by aneurysms or replaced by marginal vessels (fig 1).1,5,8
INNER RETINAL ISCHAEMIC TOPOLOGY AND RETINAL VENOUS ARTERIALISATION
In 1950, Norman Ashton42 described an Indian ink injection technique that differed in detail from that used by Michaelson. Using this protocol, Ashton illustrated, for the first time, the archetypical pattern of ischaemia in diabetes wherein broad swathes of capillary non-perfusion affect the mid-peripheral and peripheral retina (fig 4A). However, the significance of this capillary dropout, affecting >80% of the capillary net, was overlooked because the specimen was considered incompletely injected. This was despite successful filling of the entire macular capillary bed and the medium-sized arterioles and venules pursuing a meridional course between the equator and optic disc nasally and the major vascular arcades temporally. As a result, Ashton’s 1953 publication43 is generally cited instead as the first to implicate capillary closure in the pathogenesis of PDR.
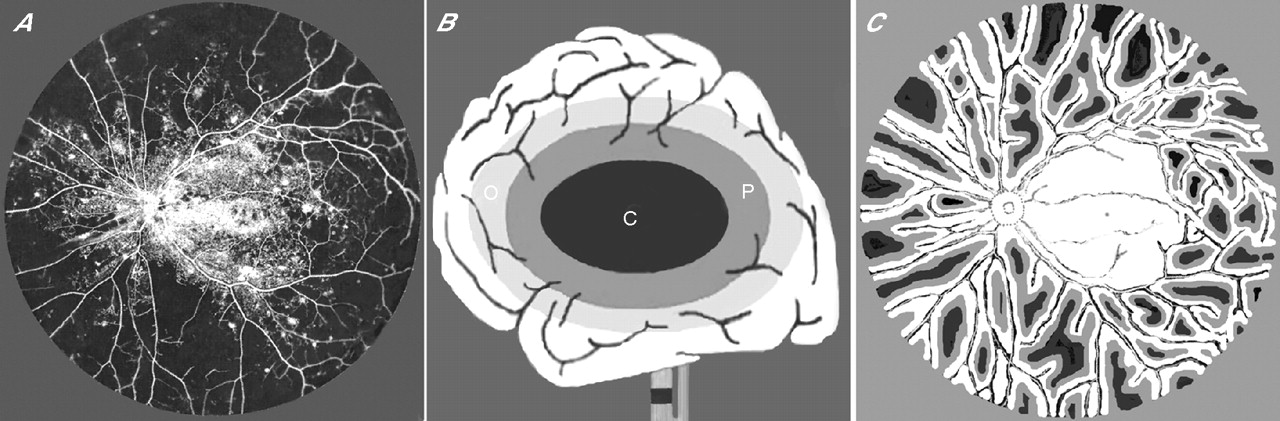
Topography of ischaemia. (A) Ashton’s 1950 Indian ink injection study42 of postmortem eye from a 71-year-old patient with diabetes of 26 years duration; illustration scanned and positive-to-negative inverted to simulate fundus fluorescein angiography. Extramacular capillary closure through 360°; medium-sized arterioles and venules radiate and anastomose across mid-peripheral retina. (B) Diagram after Kidwell et al61 showing the consequences of internal carotid artery occlusion in cerebral grey matter. Ischaemic penumbra (P) interposed between core of infarction (C) and zone of benign oligaemia (O). (C) Hypothetical map of diabetic inner retinal grey matter penumbra (based on histopathological and visual field data) superimposed on vessel topology of Ashton’s 1950 43 preparation; well-oxygenated tissue (white), penumbra (grey) and infarct (black).
Retinal digest and in vivo studies subsequently confirmed that capillary non-perfusion in diabetes varies in extent from closure of individual capillaries to obliteration of the whole capillary net apart from that serving the immediate peripapillary retina.44 It was not until 1981, however, that FFA montages from Shimizu and colleagues45 displayed the same ischaemic topology as that illustrated by Ashton 31 years earlier, and showed that this is by far the most common pattern in PDR. The oval interface between perfused and non-perfused retina is typically located just external to the temporal vascular arcades and nasal to the disc, and is penetrated by multiple arterioles and venules radiating into the ischaemic periphery. There the vessels are linked by arteriovenous anastomoses representing dilated capillary connections (“preferential channels”) and/or by de novo establishment of direct communications (“fistulae”) at arteriovenous crossings.43,45–48 Arteriovenous shunting and vascular dilatation presumably explain the modesty of the reduction in volumetric retinal blood flow seen in PDR,49 notwithstanding the gross tissue hypoperfusion, and probably cause retinal venous hypertension.
Shimizu supported Ashton’s hypothesis that the wholesale obliteration of the capillary bed reflects expansion of the periarterial capillary-free zone and coalescence of smaller ischaemic territories.43,45 Arteriolar occlusion by integrin-mediated leucocyte entrapment is gaining acceptance as the underlying mechanism.50 The extent of capillary dropout on FFA is greatest in eyes with rubeosis iridis, and greater in eyes with ODNVs than PRNVs alone.45 Such “proportionality” implies that, for an angiogenic response to be mounted, agonists such as VEGF must exceed a threshold intraocular concentration, while a sufficient density and avidity of angiogenic receptors is induced in the retinal, disc and iris vasculature. Panoramic FFA also demonstrated a topographical relationship between the posterior margin of mid-peripheral capillary closure and sites of neovascular outgrowth from veins in the posterior pole.45 Like “directionality”, such “proximity” had been much more obvious in well-circumscribed retinal ischaemia, as after branch vein occlusion.51–53 In diabetes, proximity to extensive capillary non-perfusion is a property also shared by venous coils and omega loops.54,55
MISERY PERFUSION AND PENUMBRAL MODULATION OF PRERETINAL ANGIOGENESIS
In 1956, George Wise pondered the neuroglial disturbances arising within inner retina affected by ischaemia, vasodilatation and neovascularisation.6 He surmised that a state of “relative anoxia” follows capillary or venous obstruction and that the cells secrete “factor x” as a byproduct of their altered metabolism. By contrast, cell death from “anaemic infarction” after occlusion of the central retinal artery (CRA) precludes production of angiogenic molecules, as does reperfusion of surviving cells or those involved in tissue repair. Wise then extended his “critical hypoxia hypothesis” to include retinal detachment, separation of the photoreceptors from the choroid being thought to induce subretinal neovascularisation.8,56 More recently, the onset of retinal detachment in PDR has been linked to rapidly accelerating rubeosis iridis.57
Studies of retinal VEGF elaboration using in situ hybridisation support Wise’s concept. Although VEGF message wasn’t noticeably different from normal in eyes with background retinopathy, heightened expression of VEGF mRNA was seen in parts of the ganglion cell layer and/or inner nuclear layer in eyes enucleated for rubeotic glaucoma.58,59 In eyes with retinal detachment, VEGF message was also increased in the outer nuclear layer.
Wise’s hypothesis can be reformulated by borrowing the notion of “misery perfusion” following cerebrovascular occlusion and applying it to the hypoxic hinterlands of the diabetic retina. In acute cerebral ischaemia, a core of irreversible grey matter infarction is often surrounded by a zone of hypoxic but viable tissue (the “penumbra”) that is itself surrounded by oligaemic tissue under no immediate threat of damage (fig 4B).60,61 Within the penumbra, the precarious balance of oxygen supply and consumption is reflected in metabolic downregulation but enhanced oxygen extraction from blood still circulating through the tissues (a property exploited in positron emission tomography). Here, secretion of growth factors such as VEGF and angiopoietin, and upregulation of their receptors induce both neuroprotection and angiogenesis.62–65 The penumbral neurons are “silent” but may escape infarction and recover their function if clot lysis, collateralisation revasculansation or endarterectomy enables tissue reperfusion. Otherwise, especially where the penumbra abuts the necrotic core, neuroprotection fails, cellsapoptose and the infarct expands.
Acute retinal arterial occlusions are seldom complicated by neovascularisation,6,8,66 but nonetheless they reveal a potential for tissue preservation and rescue. Thanks to oxygenation from the choroid, for example, the peripheral retina often survives CRA occlusion, allowing retention of the associated visual field or its early restoration.67–69 Reperfusion through the CRA probably contributes to this functional recovery and freedom from neovascular sequelae. When the CRA is only partially obstructed, tissue viability is also maintained posteriorly and extends peripherally as a mantle around vessels with a modicum of retained circulation.70–72 Oxygen diffusion from these vessels and from the choroid underlies the creation of a complex grey matter penumbra (GMP) surrounding a peripapillary zone of benign oligaemia. This is associated with retinal venous cyanosis and visual impairment, but remarkable functional recovery follows restoration of the inner retinal circulation. A persistent GMP and intraocular neovascularisation characterises chronic panretinal hypoperfusion from carotid occlusive disease,6,72 but rubeosis iridis and ODNVs may regress within hours of endarterectomy.73
The angiogenic response in PDR parallels that induced by chronic panretinal hypoperfusion, or evolving within a cerebral penumbra. Since there is no tissue reperfusion capability after diabetic capillary closure other than that sometimes ascribed to intraretinal microvascular abnormalities,74 sources of inner retinal oxygenation are limited to the vitreous, the choroid and blood shunting through the medium-sized radiating anastomosing vessels (RAVs). As with CRA occlusion, a marked reduction is seen in the neuronal population of the ganglion cell layer and inner nuclear layer, together with a variable degree of gliosis.75,76 However, sparing of a tissue mantle around the RAVs (fig 3A) enables some of the neurons to remain functional,76,77 and heightened VEGF message in a perivascular distribution has also been illustrated.59 This supports the hypothesis that RAVs are major determinants of GMP topography (fig 4C).
According to Davis, spontaneous regression of neovascularisation, part of the natural history of PDR, implies retinal ischaemia that is “too severe” to maintain vessel growth.1,5 Put another way, the angiogenic GMP may eventually be subsumed into the core of infarction owing to RAV occlusion and/or progressive choroidopathy. Similarly, after scatter photocoagulation, neovascular regression may follow the apoptotic demise of growth factor-secreting cells in the inner retina owing to choriocapillaris occlusion and reduced choroidal oxygenation.78 Alternatively, thermal destruction of the highly energetic photoreceptors, and the lower energy consumption of the glia that often replace them, might over-ride such influences and improve oxygen flux from the choroid.79 No concomitant neuronal rescue is evident, however, field constriction rather than functional gain following scatter photocoagulation.
Whichever route is taken in exiting from the GMP (whether via necrosis through apoptosis or rescue through reoxygenation), and whether this follows photocoagulation or occurs spontaneously over time, the outcome is the same. Neovascular regression is accompanied by downregulation of VEGF/VEGFR2 and PlGF/VEGFR1 in the retina and vitreous,25,26,38,39 reduced arterial diameters and volumetric blood flow,79 and reversal of venous beading and loop formation.5,55 Equatorial and postoral scatter laser is especially beneficial in attenuating the overall angiogenic drive,80,81 presumably by addressing the peripheral GMP sustained by the choroid.
NEOVASCULAR BUDDING AND BIOMECHANICAL ACTIVATION OF ANGIOGENESIS
In 1964 and 1970, John Dobree reported his longitudinal studies of PDR using 3-monthly fundus photography.7,82 Some of the neovascular lesions were at an early “naked” stage of development, whereas others within the same eye had regressed or fibrosed, suggesting that they were primarily responding to local retinal conditions rather than systemic changes.7 With Enid Taylor, Dobree then pinpointed sites of discrete neovascular outgrowth and showed that most PRNVs arise from retinal veins, only 20% deriving from the capillary bed. These locations had a composite distribution that was C-shaped and extramacular, sparing the temporal raphe.82 In preproliferative retinopathy, cotton wool sentinels in a similar distribution evolve proximate to the same ischaemic margin.1,72,83
Taylor and Dobree went on to demonstrate that the great majority of neovascular outgrowths are located on either the peripheral or the disc side of arteriovenous crossings, as later confirmed by FFA.82,45 They surmised that the site of venous budding might therefore reflect “a localised disturbance in haemodynamics”. This chimes with contemporary views on the pathogenesis of MVI in that, using the chick chorioallantoic membrane and other models, endothelial gene expression has been shown to be powerfully modulated by haemodynamically generated stimuli.13,84,85 The “biomechanical stress hypothesis” so derived links growth factor receptor expression in the vein walls to venous hypertension and/or flow disturbances from arteriovenous shunting.
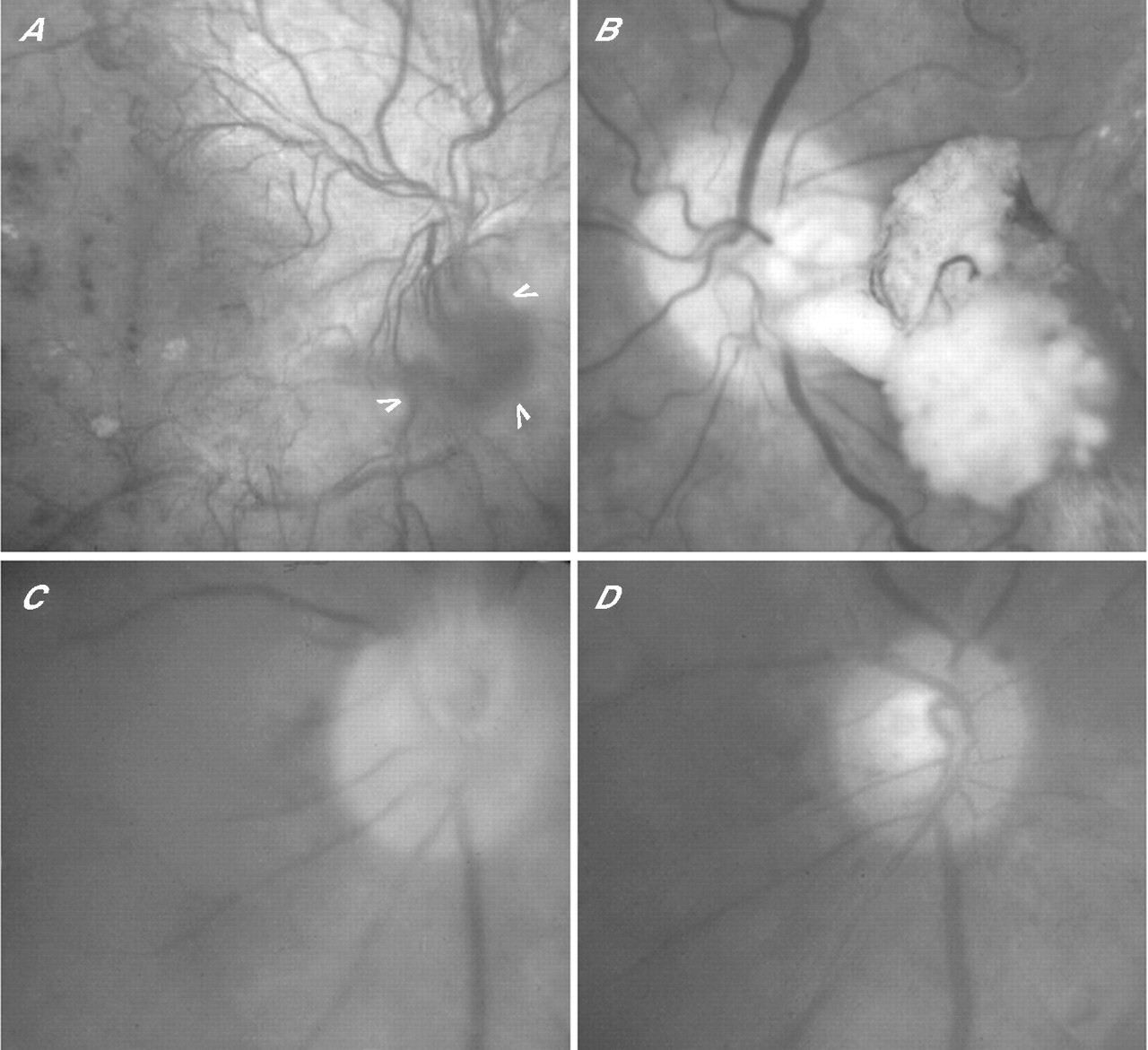
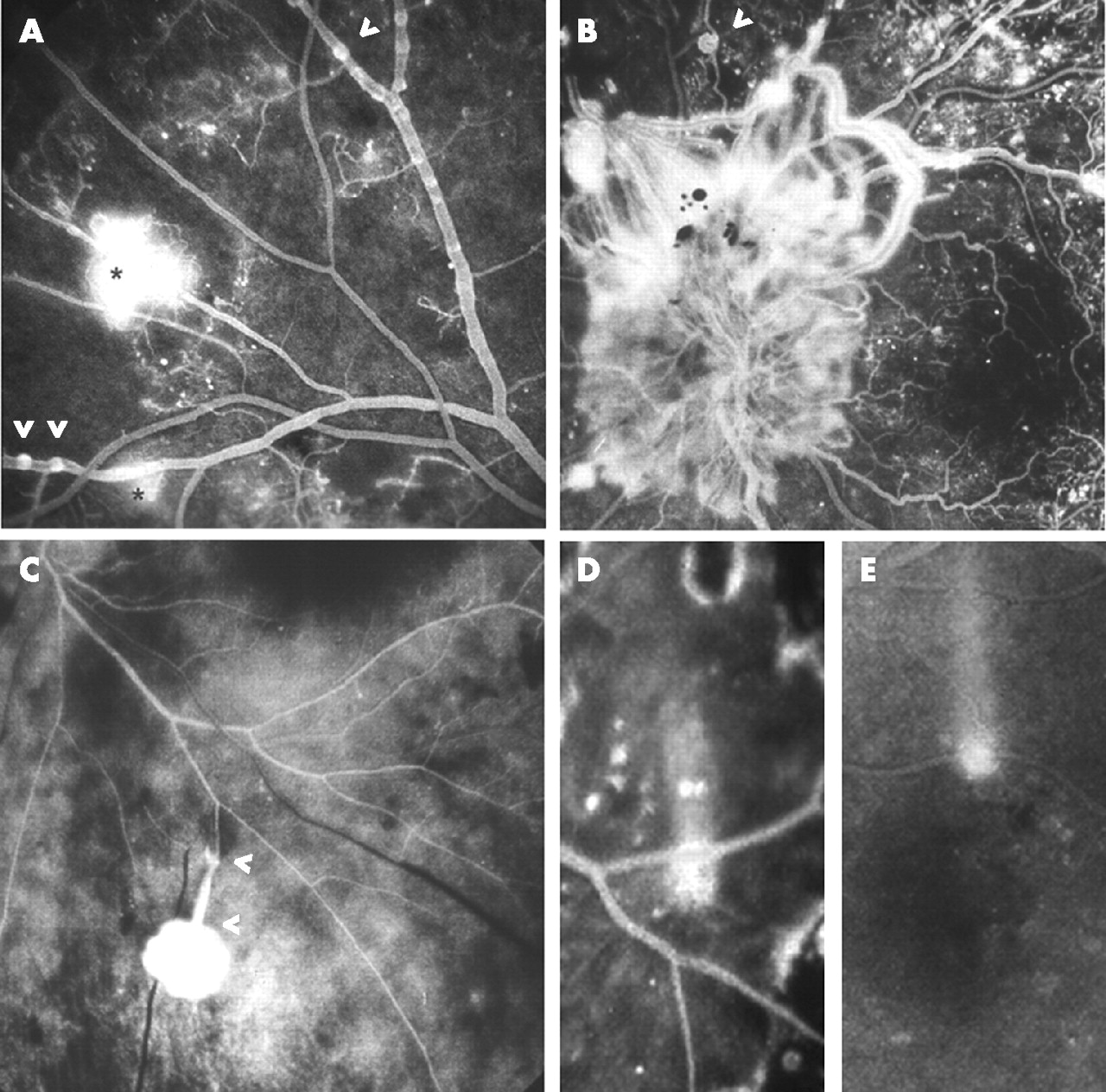
Not all eyes developing PDR manifest clinically obvious changes in the retinal veins,1 but the transparent tissue envelope around the expanded red cell column usually develops a series of nodes (venous beading), and staining is evident on FFA, mirroring Ballantyne’s phlebosclerosis (fig 5). Bleb-like buds with enhanced staining then emerge near arteriovenous crossings and/or at points of venous confluence or preferential channel insertion, and microloop formation is occasionally identifiable.8 Hyperfluorescent patches evolve thereafter, signifying neovascular endothelial modulation by the vitreous ECM once the ILL has been penetrated (fig 5A).

Fundus fluorescein angiography. (A) Against a backdrop of capillary closure, staining of venous walls reveals nodes and bleb-like buds (arrowheads) giving way to circumscribed hyperfluorescence (asterisks) indicating neovascular penetration of inner limiting lamina. (B) Delicate rete of optic disc new vessels, wide neovascular channels and a microloop (arrowhead). (C) Dye leakage from arterialised rete within detached posterior hyaloid membrane; rete connected to retina by extended peduncle traversing retrohyaloid space (between arrowheads). (D, E) Two abortive neovascular outgrowths, each showing a convectional plume of dye (D) in vitreous cavity fluid of a vitrectomised eye and (E) in retrohyaloid fluid of an eye with PVD.
Remodelling within an expanded rete mirabile can result in channels whose width exceeds that of the retinal veins, whereas similarly wide, straight vessels sometimes connect host veins to distant preretinal neovascular networks.1,5 These afferent and efferent “feeder” vessels represent extended VNPs and often originate at arteriovenous crossings (fig 1B). Bek9 reported that the topographic relationship between venous loops and arteriovenous crossings is less remarkable. However, even if such loops arise at some distance from a crossing, serial sectioning may show their interconnection through a segment of venous reduplication.86 Furthermore, distended omega loops spanning arteriovenous crossings (fig 2D′) can sometimes be shown to be arterialised on FFA,8 suggesting that MVI underpins arteriovenous fistula formation.
VITREORETINAL NEOADHESIONS AND ECM MODULATION OF ANGIOGENESIS
In 1965/6, stereoscopic studies of the natural history of PDR were reported by Matthew Davis87 and Felipe Tolentino et al.88 Using indirect ophthalmoscopy and slit-lamp biomicroscopy, they described a precise correlation between sites of neovascular projection into the vitreous cavity and the establishment of exaggerated vitreoretinal adhesions. Clinically, the earliest new vessels appeared to be sandwiched between the retina and the posterior hyaloid membrane (PHM), whose local separation from the ILL was considered important in stimulating vasoproliferation. The vessels were thought to adhere progressively to the PHM thereafter.
Subsequent histopathological studies suggested that PRNVs become “incarcerated” within attached cortical vitreous from the outset.89,90 Leaking plasma components such as fibrin and fibronectin are then added to the ECM which, like the ILL, is subjected to proteolytic modification by the expanding neovasculature. The balance of pro-angiogenic and anti-angiogenic influences therefore tips in favour of vasoproliferation through retinal overproduction and intravitreal accumulation of molecules such as VEGF, and also through sequestration or degradation of vasoinhibitory molecules in the gel. The latter include pigment epithelium-derived factor (which cleaves VEGFR1) and two molecules colocalising within the vitreous cortex, endostatin and opticin.91–94
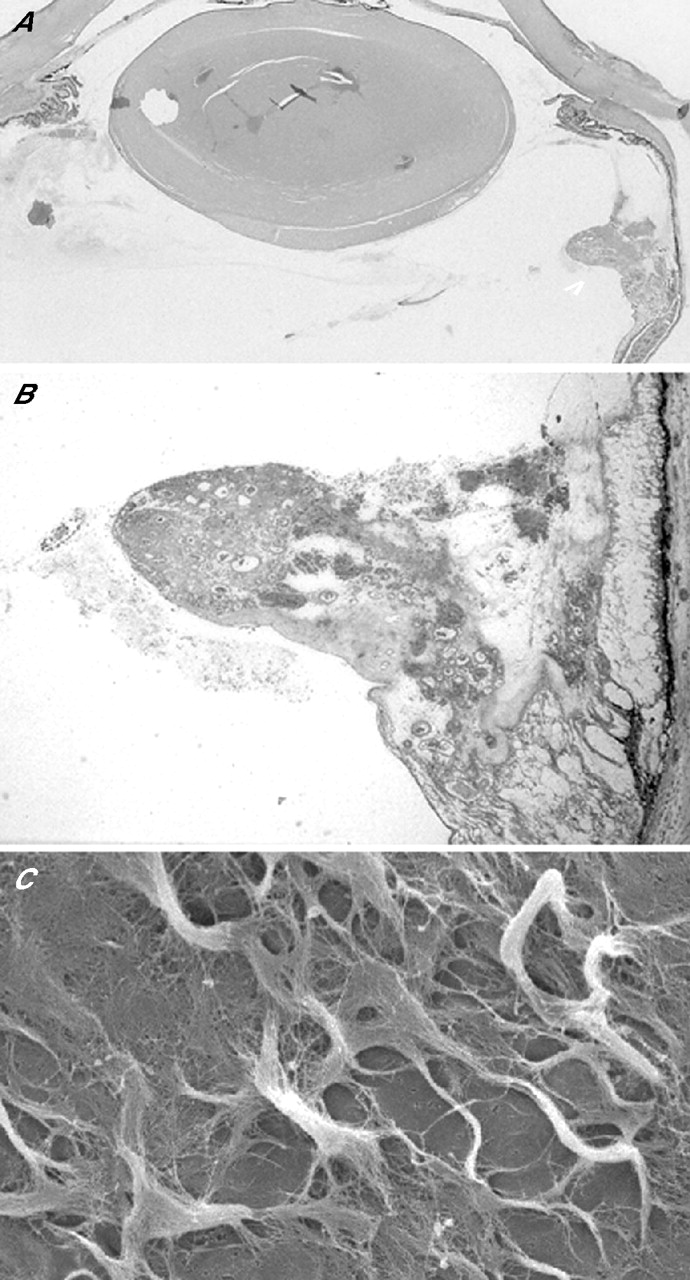
Foos95 drew parallels between the vitreoretinal neoadhesions of PDR and the normal developmental vitreopapillary adhesion, wherein glial cells at the disc margin incarcerate vitreous fibrils. An intimate horizontal conjunction indeed develops between the vascularised epiretinal membranes (ERMs) and the vitreous ECM, but vitreoretinal connectivity in PDR also depends crucially on the integrity of the VNPs. Otherwise called “vascular pegs” or “glial nails”, these structures are exposed when vertically orientated microscissors are used to “segment” a coalescent fibrovascular ERM, thus separating small tissue islands from one another and isolating the VNPs. Alternatively, horizontally orientated scissors are used to sever the VNPs—“membrane delamination” (fig 6).96 Unfortunately, en face biomicroscopic views of fully established ERMs may give little indication of the density of their peduncular retinal attachments, and therefore what surgical difficulties to anticipate.

Delamination of fibrovascular epiretinal membranes (ERMs). (A) Diagram of scissors delamination; no untoward bleeding from the cut ends of venous neovascular peduncle. (B) Diagram of viscodelamination; avulsion of the venous neovascular peduncle from the retinal vein while injected viscoelastic constrains bleeding. (C) Diagram of spontaneous PVD and peduncular avulsion; blood clot on retinal surface (after McLeod and James110; for clarity, venous neovascular peduncles are depicted as containing only one channel linking rete to retinal vein). (D) Fundus photograph of left eye of a patient with PDR taken within 2 h of onset of vitreous haemorrhage; retrohyaloid blood clot beneath and surrounding fibrovascular ERM. Shrivelled segments of ERM where peduncles have avulsed and have no rete perfusion (arrowheads).
The openings in the ILL that accommodate the VNPs also allow Muller’s glia to penetrate and gain anchorage within the fibrovascular tissue,39,75,90 sometimes forming a “pavement” over its inner surface similar to that covering simple age-related ERMs (fig 3D,E).90,95,97 This neuroectodermal component may be the foundation for the surgical impression that established ERMs are more adherent than earlier proliferations57; it may reinforce the VNP in its role as the conduit for transmitting ERM contraction forces into the sustentacular architecture of the retina, and it may contribute to the persistence of vitreoretinal adhesion even after neovascular regression.
Most vascularised vitreoretinal adhesions can resist the forces of vitreoretinal separation, and ODNVs will augment the pre-existing vitreopapillary connection. Extension of PVD is constrained by “point adhesions” or by “zonal adhesions” from single or from multiple adjacent VNPs, respectively (fig 7). The adhesions eventually become isolated from one another and from the vitreous base while remaining interconnected through the detached PHM.53,55,57,96 Associated elevation of the edges of the vascularised ERMs may be complicated by a “sudden spurt” or exacerbation of vasoproliferation along the contiguous PHM,87 but whether such “neo-neovascularisation” occurs independently of tractional retinal detachment is uncertain (fig 7A). Myofibroblasts regularly proliferate and migrate anteriorly towards the vitreous base, however, their contraction immobilising and shrinking the PHM (fig 7B). The anteroposterior vitreoretinal traction thus generated may compound the tangential forces of ERM contraction in detaching the retina, but otherwise can lead to tractional retinoschisis98,99 or tractional extraction of a “lambda (λ) vascular loop” from the retina (fig 7C).
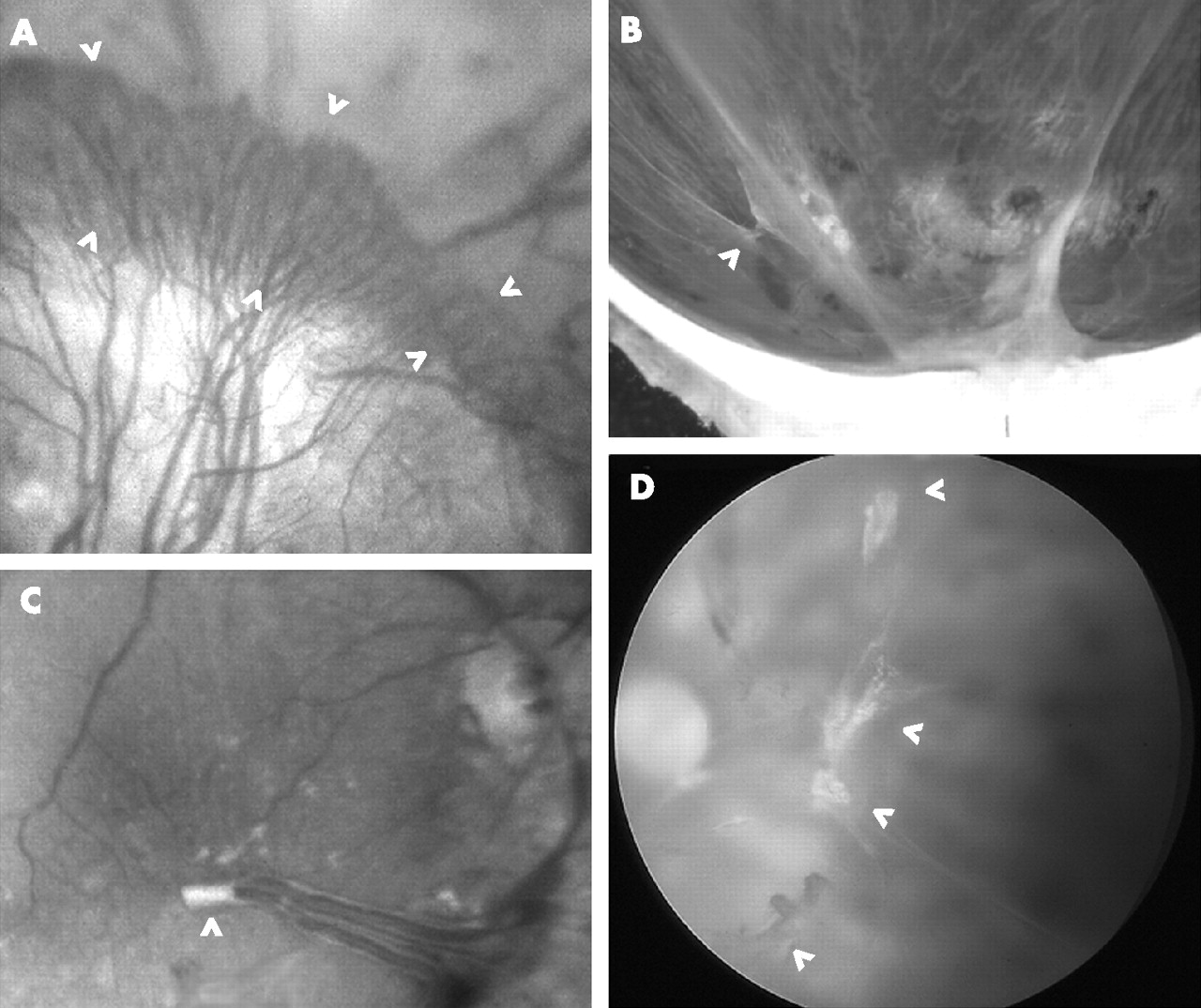
Fibrovascular epiretinal membranes (ERMs) and neovascular peduncles. (A) Brush of capillary loops (between arrowheads) proliferating along detached posterior hyaloid membrane (PHM) towards vitreous base; loops arise from edge of mature ERM causing underlying tractional retinal detachment. (B) Macrophotograph from a hemisected eye postmortem; incomplete detachment of vitreous with taut PHM; point adhesion of vitreous to retina through venous neovascular peduncles (VNP; arrowhead) and zonal adhesion over optic disc. (C) Macular retina after vitrectomy; amputated peduncle (arrowhead) at the apex of a drooping “lambda (λ) vascular loop” comprising major inferotemporal vein and artery; these vessels were earlier “extracted” from retina by anteroposterior traction exerted through the VNP, which presumably originated near the region where the vessels were crossing. (D) Arc of completely avulsed and avascular ERMs (arrowheads) within detached vitreous cortex; retrohyaloid haemorrhage partially obscures retina nasal to disc.
Realisation that the vitreous ECM is a vital scaffold for preretinal angiogenesis began to dawn once it was appreciated that “flat” new vessels grow along horizontal planes defined by the cortical vitreous lamellae and become “forward” vessels only when that part of the cortical vitreous continuum, within which they are incarcerated, separates from the retina. This notion was underlined by the observation that no postbasal neovascular tissue reproliferates over the retina after vitrectomy, and that an established complete PVD protects against preretinal rete expansion.57,100,101 The requirement for an “ECM substrate” is now believed to be a generic property of angiogenesis, underwritten by specific integrins on the endothelial cells such as αvβ3 and αvβ5.14,102–105 As well as promoting cell adhesion to structural ECM components such as collagen and fibronectin, these transmembrane proteins are crucial to functional regulation of the neovascular endothelium—for example, through activating growth factor receptors (such as VEGFR2) within the plasmalemma while interacting with the ligand (VEGF) in the ECM.
Even in the absence of a vitreous substrate, a stunted form of neovascularisation sometimes appears on the retinal surface at the posterior margin of mid-peripheral ischaemia or from arteriovenous crossings in the macula.53,106–108 These abortive neovascular outgrowths are incapable of rete expansion, but their convoluted channels, blood-filled cavities and peduncular retinal connections are consistent with MVI. The epiretinal glomeruli are distinguishable from intraretinal lesions such as intraretinal microvascular abnormalities and “abortive foveal retinal neovascularisation”74,109 by the “smoke stack” leakage of dye wafting in the vitreal fluid on FFA (fig 5D,E); rubeosis iridis generates a similar convectional plume in the aqueous.83 This contrasts with the relative confinement of leaking protein-bound dye to the immediate vicinity of new vessels growing within cortical gel (fig 5A–C). Retrosilicone oil neovascularisation is an example of preretinal angiogenesis prospering in the absence of a vitreous substrate. The confines of the retrosilicone space apparently ensure a rich concentrate of growth factors and a self-perpetuating cycle of plasma leakage and deposition of alternative angiogenic substrates such as fibrin.53,96,110,111
The limited availability of ECM substrates within the neuroretina probably explains the virtual absence of intraretinal revascularisation in PDR. However, in adults, a matting of collagen fibrils is secreted into the superficial retina at its extreme anterior limit.112,113 Provided RAVs extend to this remote location, therefore, new vessels can proliferate intraretinally before accessing the basal gel and anterior hyaloid alongside heterotypic fibrils that penetrate the postoral ILL and splice with those in the vitreous cortex (fig 8). Such “retrolental neovascularisation” or “anterior hyaloidal fibrovascular proliferation” first came to attention as a complication of vitrectomy in eyes with untreated peripheral retinal ischaemia and scleral buckling.96,114 However, it is now evident that, irrespective of PVD or vitrectomy, a limited form of basal vasoproliferation is often present in eyes with rubeosis iridis in the guise of an oral ridge, best appreciated endoscopically.115

Basal vasoproliferation. (A, B) Sections from an otherwise unoperated eye excised for diabetic rubeosis iridis (courtesy of Dr R Bonshek). (A) Oral ridge of vasoproliferation (arrowhead) with detail (B) showing intraretinal vessels extending into basal vitreous. (C) Scanning electron microscopy of the undersurface of postoral ILL from normal eye after retinal cell digestion: angiogenic substrate from sublaminar matting of skeins of heterotypic fibrils (Wang et al).112
Stefansson et al79,116 have rejected the “vitreous substrate hypothesis”, proposing instead that improved inner retinal oxygenation by fluid vitreous explains the absence of ODNVs and postbasal PRNVs after complete PVD or vitrectomy. However, although vitreous oxygenation can indeed protect the innermost ganglion-cell axons from ischaemia,72,117 the lower oxygen tension in diabetic vitreous will limit retinal oxygenation through this route.118 Moreover, the concentration of VEGF remains unchanged in the vitreous cavity after diabetic vitrectomy, so hypoxia within the inner retinal GMP presumably is not alleviated.119 Scatter endophotocoagulation is usually necessary, therefore, to counter anticipated complications (such as rubeosis iridis) arising from surgical interference with barriers to intraocular diffusion of angiogenic molecules. This is of particular concern in aphakic eyes and in phakic or pseudophakic eyes with incidental or intended breaches in the anterior hyaloid (“pseudo-aphakia”).81
VENOUS PEDUNCULAR AVULSION AND VITREOUS HAEMORRHAGE
The reports from Davis and from Schepens’ group in 1965/6 also suggested a role for PVD in precipitating vitreous haemorrhaging, purportedly through its exerting traction on “unsupported” new vessels.87,88 Then, in eyes with dehiscences in the vitreous cortex, blood from the retrohyaloid compartment might penetrate into the vitreous gel. However, this hypothetical mechanism of haemorrhage induction has several shortcomings. First, the fragile PRNVs are presumed to coexist alongside vascularised adhesions capable of withstanding the forces of PVD and of transmitting tractional forces into the retina. Secondly, Ballantyne’s histopathological observations and what is known about MVI combine to deny that PRNVs are ever “unsupported”.2,9–12 Thirdly, haemorrhaging typically occurs during the later stages of PDR when PRNVs are entombed within fibrous tissue whose contraction contributes to the initiation of PVD.5,7
Defining the mechanism of haemorrhaging in PDR is problematic because blood immediately clots in front of the retina before lysing and dispersing (fig 6C,D). However, the physical consequences of PVD are revealed during the now largely obsolete microsurgical technique of “viscodelamination”. Viscous injection between the PHM and retina elevates the ERMs and stretches the VNPs, facilitating en bloc disengagement of the vitreous cortex and ERMs from the retina.110,120 As the controlled vitreoretinal separation proceeds, however, bleeding frequently occurs albeit the haemorrhage may be confined by the transparent viscoelastic and the induced ocular hypertension (fig 6B). The source of bleeding can then be identified as side-puncturing of the vein walls as VNPs are avulsed, so it is the retinal vein (rather than the preretinal neovasculature) that bleeds. VNP avulsion also complicates other microsurgical dissection techniques, whether ill-advised “peeling” of vascularised ERMs or, unintentionally, during segmentation or delamination.
This disruptive mechanism parallels Weiss ring formation during rhegmatogenous age-related PVD,121,122 and various degrees of ERM–retinal cleavage result. As noted, partial ERM avulsion will cause flat new vessels to become forward vessels (with or without neovascular exacerbation), but no bleeding arises without VNP avulsion (fig 7B). Rarely, the ERM becomes entirely disconnected from the retina other than through feeder vessels that traverse the retrohyaloid space and maintain rete perfusion (fig 5C).1,5 Complete ERM and peduncular avulsion (fig 7D) will result in haemorrhaging from the arterialised retinal veins, and empty “ghost vessels” may then be discernible within the aerialised ERMs embedded in the detached PHM. Most frequently, however, avulsion of part of a fibrovascular ERM, including one or more VNPs, will precipitate bleeding (fig 6C,D), while only the affected ERM segments undergo autoinfarction (assuming no rete anastomosis).
Vitreous haemorrhaging in PDR is thus an inherently self-healing process. The venous bleeding point seldom leaks after puncture repair, and the non-availability of a vitreous substrate ensures that there is no further PRNV growth locally. Each haemorrhage is potentially a step towards eventual cessation of bleeding through completion of PVD or attainment of a residue of stable vitreoretinal adhesions that are not susceptible to avulsion. The absence of PRNVs or ODNVs from an eye containing a vitreous haemorrhage does not necessarily imply that PDR was not its cause (given the bleeding mechanism’s ephemeral nature), while the discovery of new vessels in an eye with a vitreous haemorrhage does not dictate that these were responsible for the bleeding. Moreover, prior regression of neovascularisation and shutdown of rete perfusion does not guarantee protection from PVD-induced VNP avulsion and vitreous haemorrhaging. This has implications for defining clinical end points when assessing antiangiogenic treatments.
PREPAPILLARY NEOVASCULARISATION AND OPTIC DISC STALK FORMATION
The reports from Dobree and Taylor (1964, 1970)7,82 also demonstrated that ODNVs are generally more extensive than PRNVs when first identified in the clinic, and they evolve more rapidly through their developmental stages. FFA later showed that eyes with ODNVs have more extensive capillary closure than those with PRNVs alone, and that the posterior margin of mid-peripheral ischaemia is closer to the disc.45 Issues of proportionality and proximity aside, however, an exaggerated responsiveness of ODNVs has been postulated and variously ascribed to deficiencies in papillary tissue barriers (for instance, the thin ILL), to posterior drainage of vitreal fluid through the disc, or to “priming” by primary hyaloid remnants.6,8,53,123 The abundance of prelaminar adventitial substrates, as well as haemodynamic factors such as the vascular waterfall within the central retinal vein (CRV) may also contribute. Occasionally, ODNVs seem to originate from the arterial supply to the optic nerve head rather than the CRV or its tributaries.55,124,125
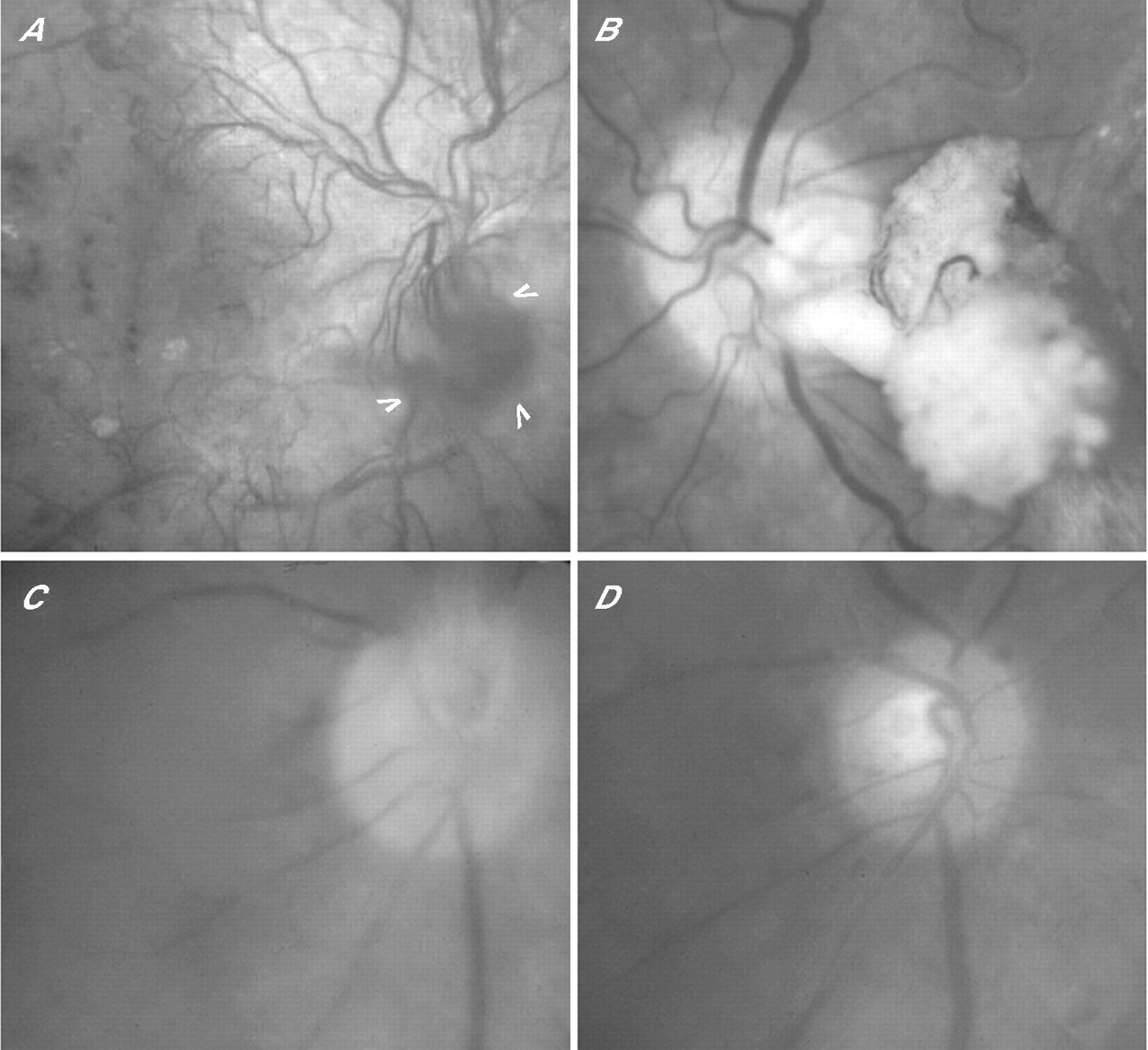
By definition, there is no secondary vitreous within the posterior expansion of Cloquet’s canal that encloses the optic disc. An epipapillary angiogenic substrate nonetheless exists, enabling ODNVs to traverse the roof of the physiological cup after emerging from the disc rim1,5,45 and also to form a rete, confined to the disc, in some eyes with complete PVD (as confirmed by smoke-stack disc fluorescence).107 Provided the vitreous remains attached to the peripapillary retina, however, ODNVs can undergo seemless, multidirectional, epiretinal extension using the secondary vitreous cortex as their substrate. Membranes containing ODNVs may then coalesce with juxtapapillary fibrovascular tissue arising from the retinal veins. Alternatively, feeder vessels with no connection to the underlying retinal vasculature supply preretinal networks that are exclusively papillary in origin (fig 1C). Vasoproliferation anteriorly along Cloquet’s canal is an occasional accompaniment (fig 9A).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fundus photographs of disc area. (A) Peripapillary fibrovascular epiretinal membrane and vascular rete (arrowheads) growing forwards along Cloquet’s canal. (B) Vitrectomised eye with fibrovascular stalk in situ arising from inferotemporal disc rim. (C, D) Anteroposterior traction on “forward optic disc new vessels (ODNVs)” forming a short stalk (C); same eye 1 week later after the clearance of intervening vitreous haemorrhage and showing complete avulsion of ODNVs from the central vein (D).
A stalk of fibrovascular tissue sometimes emanates from the disc in advanced PDR (fig 9B). Its formation can be attributed to the effects of PVD on juxtapapillary preretinal fibrovascular tissue that becomes forwardly disposed as the cortical vitreous separates from the retina. Fibrous contraction then creates a perpendicular structure with a horizontal rete at its point of attachment to the detached PHM.106,126 The amount of associated bleeding (if any) reflects the number of retinal VNPs avulsed in the process, while spontaneous avulsion of ODNVs from the CRV also occurs occasionally (fig 9C,D). This entity is not to be confused with a “residual optic disc stalk” fashioned during segmentation of coalescent preretinal and prepapillary fibrovascular membranes, and which some surgeons prefer to avulse from the CRV and remove despite the risk of major haemorrhage.127
CONCLUSIONS
The clinical definition of PDR is that of preretinal and prepapillary vascular rete formation. This has the advantage of pragmatism in predicting PDR’s haemorrhagic and tractional sequelae, but is arbitrary. Indeed, to distinguish PDR from “preproliferative” or “severe non-proliferative” retinopathy is to obscure their pathophysiological commonality.1,5 Reflecting the findings of Ballantyne (1946)2 and Taylor and Dobree (1970),82 MVI is proposed as the fundamental link between venous budding, arteriovenous fistulae, VNPs, feeder vessels, rete mirabile, abortive neovascular outgrowths, venous reduplications and omega loops.
Michaelson (1948), Ashton (1950) and then Wise (1956) in turn predicted, demonstrated and then elucidated the intraretinal vascular changes underpinning the various vasoproliferative lesions of PDR.6,18,42 By virtue of comparable gradients of hypoxia developing within the neuropile, diabetic capillary closure can be reconciled with cerebrovascular occlusions where penumbral neuronal rescue rather than angiogenic revascularisation was of prime interest until recently. Growth factor receptor upregulation in the retinal venous endothelium and its adventitia appears to be instigated by biomechanical stress as well as by the chronic GMP, and the RAVs have a role in generating both. A reduction in the volume of the GMP and neovascular regression follows tissue revitalisation by reoxygenation or, equally, tissue necrosis by apoptosis. In future, using physiological imaging to map the GMP may foster a more rational and potentially sight-enhancing approach to photocoagulation treatment.
Stimulated by the observations of Davis (1965) and Schepens’ group (1966), preretinal neovascularisation came to be recognised as being subject to the presence of an appropriate and accessible ECM substrate.87,88 Indeed, nowhere in the entire realm of clinical pathology is the importance of integrin-dependent vascular cell adhesion more clearly evident than at the postbasal vitreoretinal juncture. The virtual absence of retinal revascularisation after capillary closure, and variations in angiogenetic responses in the vitreous base and optic nerve head, are similarly explained. When the vitreous ECM detaches, however, the origins of the VNPs from the retinal veins are exposed as the points of greatest mechanical weakness within the neovasculature.
Acknowledgments
I thank JF Cullen, EM Kohner, RK Blach, PK Leaver, J Marshall, I Grierson, ME Boulton and PN Bishop for their inspiration and interaction in the field of diabetic eye disease. Grant support from The Wellcome Trust, British Diabetic Association, Guide Dogs for the Blind Association, Ross Foundation (Scotland) and the research endowment funds of Moorfields Eye Hospital and the Manchester Royal Eye Hospital is gratefully acknowledged.
REFERENCES
Footnotes
-
This is an update of the Duke Elder Lecture to the Annual Congress of the Royal College of Ophthalmologists for 1993.
-
Competing interests: None declared.
Linked Articles
- Correction