Article Text

Abstract
Age-related macular degeneration (AMD) is still referred to as the leading cause of severe and irreversible visual loss world-wide. The disease has a profound effect on quality of life of affected individuals and represents a major socioeconomic challenge for societies due to the exponential increase in life expectancy and environmental risks. Advances in medical research have identified vascular endothelial growth factor (VEGF) as an important pathophysiological player in neovascular AMD and intraocular inhibition of VEGF as one of the most efficient therapies in medicine. The wide introduction of anti-VEGF therapy has led to an overwhelming improvement in the prognosis of patients affected by neovascular AMD, allowing recovery and maintenance of visual function in the vast majority of patients. However, the therapeutic benefit is accompanied by significant economic investments, unresolved medicolegal debates about the use of off-label substances and overwhelming problems in large population management. The burden of disease has turned into a burden of care with a dissociation of scientific advances and real-world clinical performance. Simultaneously, ground-breaking innovations in diagnostic technologies, such as optical coherence tomography, allows unprecedented high-resolution visualisation of disease morphology and provides a promising horizon for early disease detection and efficient therapeutic follow-up. However, definite conclusions from morphologic parameters are still lacking, and valid biomarkers have yet to be identified to provide a practical base for disease management. The European Society of Retina Specialists offers expert guidance for diagnostic and therapeutic management of neovascular AMD supporting healthcare givers and doctors in providing the best state-of-the-art care to their patients.
Trial registration number NCT01318941.
- Retina
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Age-related macular degeneration (AMD) has been described as the leading cause of legal blindness, affecting 10%–13% of adults over 65 years of age in North America, Europe, Australia and, recently, Asia.1 ,2 AMD is a major medical and socioeconomic challenge worldwide and, based on increased life expectancy and a growing negative impact of environmental risk factors, particularly arteriosclerosis, obesity and smoking, its incidence is expected to at least double by 2020.3–7 The Global Burden of Disease Study 2010 reported an exponential increase of 160% in vision-related years lived with disability due to AMD, highlighting the overwhelming burden for societies overall.8 Moreover, with an impact similar to AIDS, renal failure and stroke, AMD has a profound effect on the quality of life of those affected.9
Fortunately, progress in AMD's diagnosis and therapy, based on advances in medical research has recently wrought a substantial paradigm shift in the management of neovascular AMD. Identification of a major pathogenetic feature, that is, the influence of vascular endothelial growth factor (VEGF), has opened an easily accessible therapeutic window.10–12 Landmark clinical trials proved that intravitreal inhibition of VEGF-A can efficiently block the pathophysiological process of AMD, restore retinal morphology and increase/maintain neurosensory function in most patients with neovascular AMD.13 ,14 Since the approval of anti-VEGF pharmacotherapy in 2006, the prevalence of legal blindness and visual impairment due to AMD has been considerably reduced, removing neovascular AMD from the list of incurable diseases.15–17
The impressive benefit of antiangiogenic therapy has since been widely recognised. However, real-life outcomes have consistently been found to be less favourable than clinical trial results.18–20 The community faces a huge dilemma in the management of AMD, with substantial controversies over the efficacy of substances, choice of therapeutic regimens and adequate monitoring needs. This is further aggravated by exponentially growing costs resulting from highly priced drugs, increasing patient numbers and long-term disease chronicity.21 ,22 At the same time, one of the most successful therapeutic break-throughs in ophthalmology is currently at the centre of an array, of unresolved issues and the standard-of-care is vastly inconsistent.
Evidently, there are enormous variations in clinical practice, and considerable uncertainty about how the scientific evidence should be reduced to clinical practice in widely varying settings. The EURETINA community has, therefore, taken responsibility for bringing together experts in the field to develop a working guidance based on the best available scientific and clinical knowledge. The goal is to provide clinically sound, economically acceptable and unbiased diagnostic and therapeutic recommendations to brighten the horizon for patients and physicians worldwide.
Diagnostic procedures
Patients’ history, clinical examination and self-monitoring
Rationale
Neovascular AMD is an acute onset and rapidly progressing disease which impacts central vision. Early detection of disease onset and continuous follow-up are mandatory because, visual loss becomes irreversible with delayed diagnosis and treatment. General ophthalmologic examination procedures, such as determination of best-corrected visual acuity (BCVA), stereoscopic ophthalmoscopy and home monitoring between routine visits should be implemented. Whenever neovascular AMD is suspected, advanced diagnostic measures such as fluorescein angiography (FA) and optical coherence tomography (OCT) must follow to confirm the diagnosis. Numerous clinical trials have shown that better final outcomes can be achieved with better initial visual acuity (VA). Unfortunately, in most trials as well as in real life, lesions nowadays are usually detected when there is already considerable visual loss. Therefore, awareness must be raised in individuals aged 50 years and older, and physicians should perform AMD screenings regularly.
Evidence
The reduced efficacy of anti-VEGF therapy compared with academic trial results is commonly associated with poor initial diagnosis and/or discontinuous follow-up in routine clinical care. Compared with treatment paradigms validated by clinical trials, patients with neovascular AMD received too few injections and only infrequent monitoring in US clinical practice from 2006 to mid-2011. Holecamp et al, found 8767 patients were treated with a mean annual number of 4.7 bevacizumab or 5.0 ranibizumab injections between 2006 and 2007. The mean annual number of injections increased slightly from 2008 to 2010, with 10 259 patients divided between six cohorts receiving 4.6, 5.1 and 5.5, bevacizumab or 6.1, 6.6 and 6.9, ranibizumab injections, but mean numbers of visits to an ophthalmologist and OCT examinations remained low.23 In a Germany-based, multicentre, retrospective review of data from patients with suspected neovascular AMD visiting ophthalmology clinics over 18 months in 2008–2010, 10 sites collected data from 2498 patients with a mean VA of 0.4±0.3 at the time of diagnosis. The most frequent pathological findings detected by routine ophthalmic examination were fibrotic lesions, indicating late diagnosis of choroidal neovascularisation (CNV).24 A confirmed diagnosis of neovascular AMD was most frequently based on funduscopy (67.3%) or FA (39.6%).
Disease activity in neovascular AMD is lifelong. Long-term outcomes 7–8 years after initiation of intensive ranibizumab therapy were assessed in patients originally treated with ranibizumab in landmark phase 3 trials (SEVEN-UP). Approximately 7 years after initiation of ranibizumab therapy in the ANCHOR or MARINA trials, one-third of patients had good visual outcomes, whereas another third had poor outcomes.25 Compared with baseline, almost half the eyes were stable, whereas one-third had declined by 15 letters or more. Hence, even at this late stage in the therapeutic course, exudative AMD patients remain at risk for substantial additional visual decline. Active exudative disease was detected by spectral-domain OCT in 68% of study eyes, and 46% were receiving ongoing intraocular anti-VEGF treatments.
The AMD Detection of Onset of New Choroidal Neovascularisation Study (AMD DOC Study) evaluated the sensitivity of time-domain (TD) OCT relative to FA, in detecting new-onset neovascular AMD within 2 years from onset. The sensitivity of Amsler grid testing, preferential hyperacuity perimetry (PHP), OCT, and FA for detection of CNV was 0.40 for OCT ((95% CI (0.16 to 0.68)), 0.42 for supervised Amsler grid (95% CI 0.15 to 0.72) and 0.50 for PHP (95% CI 0.23 to 0.77)). The AMD DOC Study demonstrated that FA is still the best method for detecting new-onset CNV.26 Nevertheless, self-monitoring with regular Amsler grid testing is suggested between ophthalmological visits. PHP telemonitoring is a more standardised self-monitoring tool. The HOME study, a prospective, randomised clinical trial found that participants randomised to the home monitor had less vision loss at the time of CNV detection than those in standard care with about 90% maintaining a VAof 20/40 or better at the time of CNV detection.27 The study also showed that when using the home monitoring device with standard care, CNV detection rates increased statistically significantly, and with smaller lesion size VA at detection was better than standard care alone. With subjective symptom realisation, BCVA at the time of detection was statistically significantly worse than an alert by the device, with a –11.5 letter loss. Increased intraocular pressure (IOP) is another issue in prolonged anti-VEGF therapy.28 ,29 In 528 eyes receiving 1796 intravitreal injections of bevacizumab, IOP was persistently elevated in 19 eyes (3.6%, 19/528) of 18 patients (4.2%, 18/424) with IOP elevated 30–70 mm Hg, 3–30 days after injection. Mean IOP was 42.6 mm Hg (range 30–70); IOP elevations occurred after an average of 7.8 injections of bevacizumab (range 3–13). Injected eyes (19/528) had a significantly higher incidence of elevated IOP than non-injected eyes (fellow eyes), 1/328, p<0.001. Identical observations were published for IOP increases with ranibizumab.29 Doctors should be aware that IOP might increase after repeated treatments.
Recommendation
Doctors should initially ask patients who present with an onset of decreased vision or metamorphopsia, if they have a family history of AMD, and for their social history including smoking habits. Their complete history should be examined to identify systemic risk factors for anti-VEGF therapy and current medications. Standardised BCVA testing and stereoscopic biomicroscopy/ophthalmoscopy of the macula of both eyes is mandatory, as well as measurement of IOP at least every 6 months. Patients should be instructed to self-monitor their vision between routine office visits. By contrast with current home monitoring strategies, those with intermediate AMD (large drusen in one or both eyes) could benefit from home monitoring with PHP, whenever the device is available. Patients who have received treatment should be monitored at regular intervals, according to a standardised strategy, either monthly or following an individualised pro-re-nata (PRN) or treat-and-extend regimen. Follow-up visits should include examination for new onset of a decrease in vision and nes or persistent metamorphopsia, changes in medical or social history and, most importantly, BCVA tests should be repeated using identical procedures. Further examination by OCT is required if stereoscopic fundus examination reveals clinical signs of retinal oedema, detachment of the retinal pigment epithelium (RPE) or haemorrhage. These recommendations are based on the Age-Related Eye Disease Study and HOME study (evidence level I) and levels II/III data for clinical management of early AMD.
Angiography
Rationale
FA was the main, and for many years, the only diagnostic and follow-up tool for AMD patients.30 Nowadays, many non-invasive techniques (such as spectral domain (SD) OCT, autofluorescence imaging) can provide detailed anatomical information and precise functional data. In spite of this, FA continues to play a key role in the diagnostic process, for example, providing the base for its clinical classification and the initiation of therapeutic management. The role of FA is to visualise retinal vasculature and neovascular retinal/choroidal proliferations as well as its dynamic features, such as perfusion and exudation. FA has been used in all phase 3 clinical trials for the initial diagnosis of neovascular AMD.
Evidence
In the case of neovascular AMD, leakage of dye from pathological new vessels, into retinal structures appears as hyperfluorescence, which increases in intensity and extension throughout the examination duration.30 This leakage is classified by its location (subfoveal, juxtafoveal, or extrafoveal) and by its features (classic, occult, or mixed). Classic CNV represents a lesion that has penetrated the RPE layer and is located in the subretinal space (figure 1), whereas occult CNV refers to a neovascular lesion located underneath the RPE (figure 2). In the case of dry AMD, the angiogram will show various grades of drusen (usually seen as early, intensely hyperfluorescence spots) and atrophy (a well-demarcated, hyperfluorescent areas resulting from increased visualisation of the adjacent choroidal fluorescence).

Classic choroidal neovascularisation is located above, the retinal pigment epithelium layer and is associated with intraretinal cystoid spaces and/or subretinal fluid. Due to its subretinal location, the neovascular net is delineated with distinct margins. Leakage in late-phase angiography confirms the biologic activity of the lesion (ophthalmoscopy, spectral domain-optical coherence tomography, early fluorescein angiography (FA), late FA).
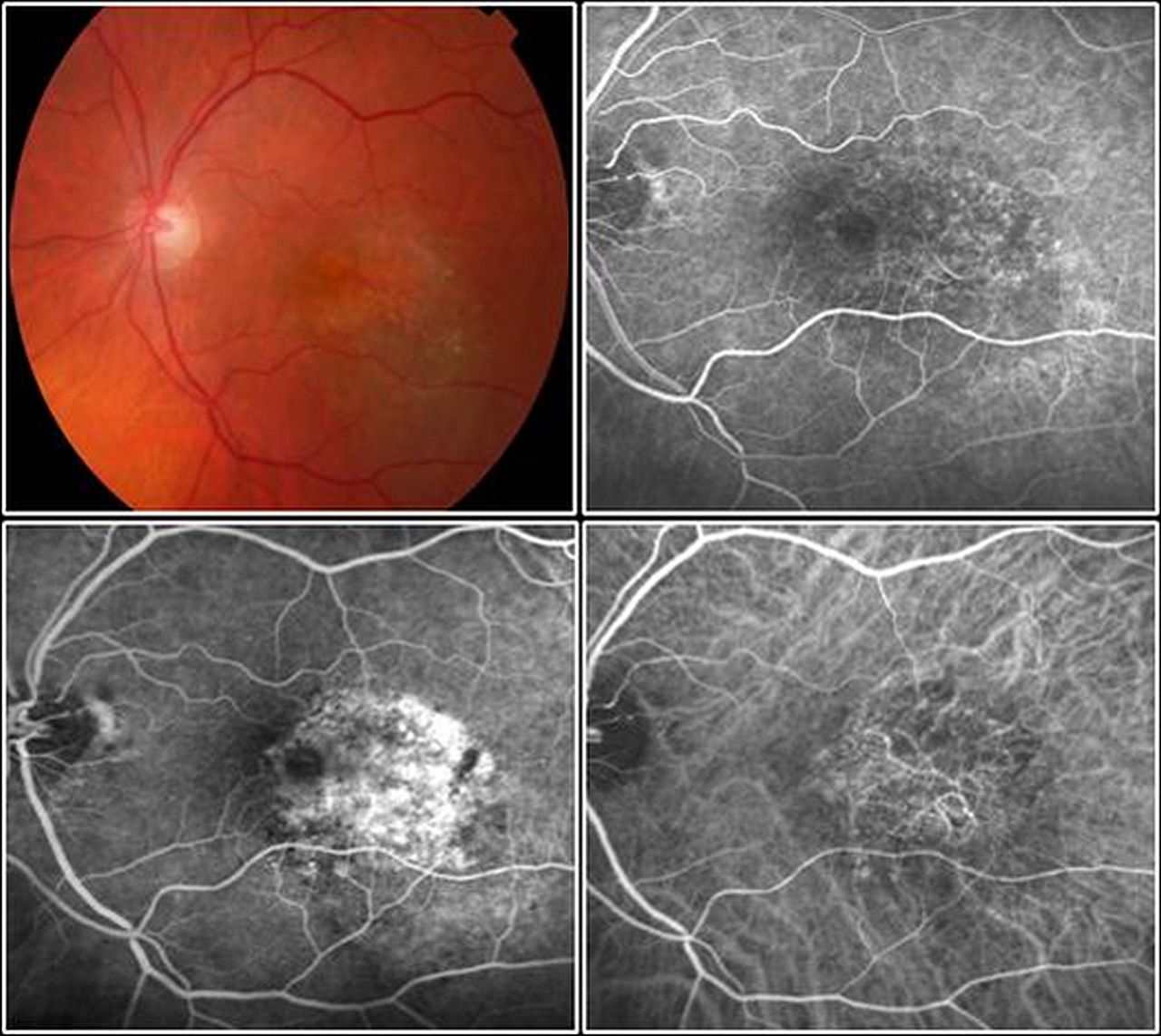
Occult choroidal neovascularisation is located underneath the retinal pigment epithelium layer. By fluorescein angiography (FA), an area of stippled, or pinpoint hyperfluorescence with leakage in late phases, are seen. Indocyanine green angiography (ICGA) (right lower image) may visualise the neovascular pattern of the occult lesion (ophthalmoscopy, early FA, late FA, ICGA).
When assessing a patient with clinical suspicion of neovascular AMD, FA evaluation, if not contraindicated for systemic risks, is routinely mandatory. 26 ,31 In fact, it is the only examination that can confirm the mere existence of a CNV, and is also used to evaluate the location and extent of classic and occult forms, particularly when it is coupled with indocyanine green angiography (ICGA). In addition to the location and the area of leakage, FA provides information about the dynamic exudative activity of the lesion. These features, particularly lesion size, have a well-recognised prognostic value and should be clarified in order to plan an appropriate treatment strategy.32–34
An angiogram is also essential to detect specific forms of AMD that present a more aggressive natural history and requires modification of the therapeutic approach. Retinal angiomatous proliferation (RAP) is characterised clinically by focal haemorrhage, oedema and lipid exudates within retinal layers. In more advanced stages, a serous or vascularised pigment epithelial detachment (PED) is detectable.35 ICGA reveals the area of focal hyperfluorescence arising from the deep capillary plexus forming the initial angiomatous lesion, which subsequently will form an anastomosis with the choroidal circulation (figure 3). ICGA is therefore vital to distinguish this lesion presentation and should be followed by SD-OCT focused on the lesion site. The other relevant example of a different subtype of exudative AMD is polypoidal choroidal vasculopathy (PCV). It is difficult to distinguish this entity clinically from other forms of occult CNV, even though, it presents more commonly with recurrent serous and haemorrhagic PED.36 ,37 The FA shows an ill-defined occult leakage pattern, whereas ICGA is able to delineate the polypoidal lesions in distinct detail (figure 4). As PCV is more common in patients of Asian and African descent, it should be considered in these patients.

A retinal angiomatous proliferation is characterised by an early hyperfluorescent spot at the level of the retinal vasculature, mostly at the site of a focal haemorrhage and progressive intraretinal leakage. The concomitant optical coherence tomography scan reveals a pigment epithelium detachment and intraretinal cystoid expansions.

Marked intraretinal exudates and/or haemorrhage seen clinically are associated with multiple hyperfluorescent polyps angiographically in polypoidal chorioidopathy. Indocyanine green angiography (ICGA) is often helpful in delineating the polypoidal components despite haemorrhage (ophthalmoscopy, early fluorescein angiography (FA), ICGA, late FA).
Recommendation
Once the initial diagnosis of CNV is established by FA, the effect of anti-VEGF therapy can be efficiently monitored by non-invasive SD-OCT alone.38–41 Nonetheless, FA may be advisable, especially where OCT fails to provide reliable information, such as in high myopia, extrafoveal lesions or when dealing with fresh CNV reactivation at the borders of a fibrotic lesion. Additionally, FA and ICGA should be repeated in the case of a sudden clinical worsening, or in occurrence of haemorrhage or new PED. These recommendations are based on evidence levels II/III.
Optical coherence tomography
Rationale
OCT, first used in the 1990s, is based on the properties of light waves reflected from and scattered by ocular tissues, which allows anatomical changes associated with exudative AMD to be visualised and measured.42 ,43 Since its introduction with the initial time-domain technology, the modality has continued to improve, with high-definition SD technology and swept source (SS) OCT, achieving greater resolution, repeatability and applicability than earlier OCT devices.44 ,45 Advanced OCT permits high-speed retina scanning that allows complete coverage of the macular area and generation of three-dimensional retinal images. Within a few years, of its introduction, OCT became a major element in both initial diagnosis and management of patients with exudative AMD. TD-OCT has been used in most of the phase 3 clinical trials for antiangiogenic therapy in AMD either as a second outcome examination for central retinal thickness (CRT) or for retreatment indications in trials with a flexible regimen. SD-OCT has so far been used exclusively in the HARBOR study comparing ranibizumab therapy in a fixed monthly and a flexible PRN regimen. OCT visualises structural changes of the retina and RPE as a high-resolution optical ‘histology’, in a static mode, however, without identification of vascular features or any representation of dynamic processes such as perfusion or leakage.
Evidence
OCT supports the diagnosis of exudative AMD at initial presentation. Type 1 CNV (also called occult CNV) may have several manifestations in OCT (figure 5): The neovascular membrane is localised behind the RPE, creating a vascularised fibrovascular or serous PED. Subretinal fluid (SRF) presents as a dark virtual space between the retina and the RPE, often with disruption of the external limiting membrane-photoreceptor complex in the outer retina. Intraretinal exudation appears as round, dark, cystoid spaces within the retinal layers, but not all cystoid spaces are exudative features. Persistent cystoid spaces mostly have an irreversible degenerative nature. Pigment epithelium detachments are characterised by elevations of the RPE (figure 6). Serous PED present as a smooth regular and sharply demarcated, dome-shaped hyporeflective RPE elevation, whereas fibrovascular PED appears to be filled with solid layers of material of medium or high reflectivity, separated by hyporeflective clefts. On OCT, RPE tears are typically seen as a discontinuity in a large PED, with the free edge of the RPE often curled under the PED. Type 2 CNV (also called classic CNV) is localised in the subretinal space (figure 7). Most eyes with type 2 CNV present a small ‘discrete’ PED associated with the highly reflective subretinal lesions (mainly located beneath the subretinal lesion). Increased thickness of the retina, SRF, cystoid spaces and PED are commonly observed.46 RAP (also called type 3 CNV) is described as small erosion or elevated RPE, a flap sign, or, later, a focal funnel-shaped defect in the RPE, called ‘kissing sign’, accompanied by subretinal and/or intraretinal fluid (figure 8).47 In PCV, the branching vascular network appears as RPE elevations, while the polypoidal lesions appear as sharper, dome-shaped protuberances, often associated with exudative findings (figure 9).

Spectral domain-optical coherence tomography (SD-OCT) reveals a fibrovascular pigment epithelial detachment and a serous retinal detachment in a patient with age-related macular degeneration affected by a type 1 choroidal neovascularisation (scanning laser ophthalmoscopy, SD-OCT).
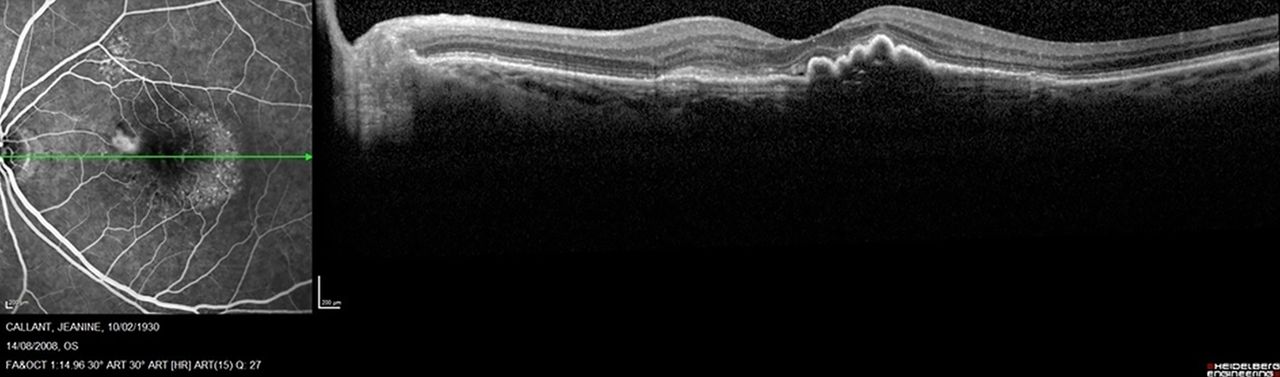
Fluorescein angiography (FA) and spectral domain-optical coherence tomography (SD-OCT) identify a minimally classic choroidal neovascularisation with the classic component in the nasal portion of the macular area and the occult component in the temporal area (FA, SD-OCT).

Spectral domain-optical coherence tomography (SD-OCT) features of type 2 (classic) choroidal neovascularisation (CNV) associated with exudative age-related macular degeneration are shown: fluorescein angiography (FA) visualises a small type 2 neovascular membrane. On SD-OCT, CNV appears between the retina and the retinal pigment epithelium, associated with some exudative cystoid spaces and increased central retinal thickness. (FA, SD-OCT).

In retinal angiomatous proliferation, fluorescein angiography (FA) shows a hot-spot in the macular area. On spectral domain-optical coherence tomography (SD-OCT), a focal pigment epithelial detachment and intraretinal cystoid spaces are the pathognomonic features. (FA, SD-OCT).

Spectral domain-optical coherence tomography (SD-OCT) features of polypoidal choroidopathy are shown: Indocyanine green angiography (ICGA) identifies a hyperfluorescent polypoidal lesion. A punctuate haemorrhage associated with the hot-spot on angiography suggests a retinal angiomatous proliferation. SD-OCT shows a dome-shaped elevation, the sign of a polypoidal lesion. (ICGA, scanning laser ophthalmoscopy, SD-OCT).
OCT is currently the most frequently used tool in the long-term management of exudative AMD. Comparisons of macular thickness and morphology over time allow a patient's response to treatment to be assessed. In the MARINA and ANCHOR studies, anti-VEGF intravitreal injections were based on a fixed regimen every 4 weeks13 ,14 and CRT measured by OCT was only a secondary outcome. Subsequently, individualised regimens based on the concept of treating patients only when necessary have since been investigated. Most subsequent clinical trials of anti-VEGF agents have used some variation on a PRN regimen, usually involving three consecutive monthly loading injections followed by further injections as needed, according to predefined retreatment criteria.48–51 This concept of individualised or evaluation-based, as-needed therapy is reportedly the most commonly used treatment regimen in current clinical practice in Europe. The most frequent morphologic criterion for retreatment decisions has been defined as an increase in CRT.52 Recent analyses revealed that CRT does not correlate with BCVA in AMD, because the structure/function correlation is lost during follow-up as early as at month 3.53 The Comparison of Age-Related Macular Degeneration Treatments Trials (CATT) study, therefore, suggested patients should be retreated in a ‘no tolerance’ mode, that is, whenever any fluid was seen on TD-OCT.54 The same principle of tight retreatment based on any change in OCT was adopted in the HARBOR trial, but, using SD-OCT which usually leads to a higher retreatment frequency due to the increased number of scans potentially revealing intraretinal fluid or SRF.55 A comprehensive subgroup analysis of the VIEW study correlation of functional and anatomical data revealed that OCT biomarkers, which are generally correlated with reduced vision in neovascular AMD, were intraretinal cystoid spaces (IRC) at baseline, and persistent cystoid spaces at the end of the loading dose.56 Whenever IRC were present initially, BCVA, and the therapeutical gain in BCVA were limited, while eyes with SRF showed the best visual prognosis. Prognostic for the therapeutic benefit were IRC and fibrovascular PED at initial presentation, where RPE detachment is the primary pathognomonic feature, and secondary cyst formation under discontinued treatment is the biomarker associated with vision loss.56 These features were independent of the substance and the regimen used.
Recommendation
BCVA alone is insufficient to detect a recurrence of activity of the neovascular membranes in neovascular AMD. FA can also be useful, in addition to OCT, in some ambiguous cases, particularly for type 2 CNV. New haemorrhage on fundus examination is also a sign of CNV activity. Nevertheless, OCT is actually the most useful tool for evaluating morphological changes because it best reflects recurrence of neovascular activity. Two types of assessment for neovascular activity can be distinguished: measurements and qualitative OCT observations.57 CRT has been the most common measurement used in clinical studies, however, PRN treatment based on these measurements was invariably associated with reduced therapeutical benefit compared with a fixed continuous regimen.54 ,58 There is a large body of evidence that supports qualitative morphology-based OCT data as more sensitive than measurements for detecting of CNV activity. IRC, SRF and RPE detachments are important signs of activity in the neovascular membrane, independent of CRT. In a ‘real life’ PRN protocol, all these features are usually considered as criteria for reinjection of anti-VEGF substances. Compared with the former TD-OCT technology, current SD-OCT or SS-OCT technologies which provide raster-scanning imaging, are more sensitive for detecting of subtle morphological changes and, thus, permit early treatment of exudative recurrence.59 ,60 The common recommendation is, therefore, to monitor disease activity using SD-OCT, and on a monthly base. The concept of a ‘zero tolerance’ on OCT criteria is emerging, because of the rapid progression of exudative features and progressive loss of vision when initiation of treatment is delayed.61 However, persistent IRC should be considered signs of irreversible retinal degeneration and should not trigger further retreatment. These recommendations are based on evidence levels I (CATT, VIEW, HARBOR) and evidence levels II.
Therapeutic strategies
Intravitreal pharmacotherapy
Pegaptanib
Rationale
VEGF increases vascular permeability, enhances the inflammatory response and induces angiogenesis.62 The isoform VEGF 165 has been particularly implicated in blood–retinal barrier breakdown and pathological intraocular neovascularisation. Pegaptanib sodium, Macugen, is a short RNA oligonucleotide, an aptamer that binds with high specificity and affinity only to the isoform VEGF165. The rationale is to selectively inhibit pathological leakage and angiogenesis.63 Pegaptanib is well tolerated in humans and has a mean intravitreal half-life of 10 days.
Evidence
The VEGF Inhibition Study in Ocular Neovascularisation (VISION) Study completed at the end of 2004 marked a new era for the treatment of neovascular AMD. The study showed the safety and efficacy of intravitreal inhibition of VEGF for the treatment of neovascular AMD over 1 year.64 It was a double-masked, sham-controlled, dose-ranging phase 3 clinical trial including 0.3, 0.1 and 3.0 mg doses. The control group was given the usual care including photodynamic therapy (PDT) monotherapy. Patients received intravitreal injections at 6-week intervals independent of the neovascular activity. Seventy per cent of those receiving pegaptanib at 0.3 mg lost fewer than 15 letters of VA, compared with 55% of the control group (p<0.001). The risk of severe loss of VA (loss of 30 letters or more) was reduced from 22% in the sham-injection group to 10% in the group receiving 0.3 mg of pegaptanib (p<0.001)(3). Patients receiving pegaptanib lost a mean of −9 letters over 1 year compared with a loss of −14 letters in the sham-injection group. Despite continued treatment, progressive growth and persistent leakage of the neovascular lesion was seen angiographically in most of the patients. At 54 weeks, every patient in the pegaptanib groups was re-randomised to continuous further pegaptanib treatment or sham treatment. Patients from the usual care group were assigned to continuous usual care, one of the three groups of pegaptanib doses or the sham-injection group. At 2 years, 59% of eyes receiving a dose of 0.3 mg lost <15 letters versus 45% of the usual care sham-injected eyes.65 Six per cent of eyes in the pegaptanib group improved by three lines compared with 2% of the sham-injected group. At 2 years, a mean vision loss of −10 letters was found in all subgroups treated with pegaptanib at 0.3 mg, with a mean of nine treatments applied per year.
The ocular and systemic safety profile of the drug was very good. Related to the intravitreal injection procedure, specific risks, such as endophthalmitis (1.3% of treated cases during the first year, 0.7% during the second year), traumatic lens injury (0.6% during the first year, 0.2% during the second year) and retinal detachment (0.7% during the first year, 1.2% during the second year) were reported.66 This was the first evidence that anti-VEGF therapy is effective and safe in AMD.
Recommendation
Macugen was approved for all lesion types in neovascular AMD by the Food and Drug Administration (FDA) for the USA in December 2004, and by the European Medicines Agency for countries in the European Union (EU) in January 2006. The therapeutic benefit was favourable compared with the one obtained with PDT monotherapy, with few treatments needed with PDT. The chance of a statistically significant improvement in VA was relatively low (6%). The visual benefits of preventing visual loss shown in the VISION Study were largely exceeded by the next anti-VEGF therapy; that is, ranibizumab. This marked difference in efficacy may be because pegaptanib does not inhibit other bioactive isoforms of VEGF, such as the soluble 110 and 121 VEGF fragment in vivo. Therefore, due to its poorer efficacy compared with other currently available anti-VEGF drugs, pegaptanib is no longer recommended for the treatment of exudative AMD. These recommendations are based on the VISION study data (evidence level I).66
Ranibizumab
Rationale
Ranibizumab is a recombinant, humanised Fab fragment of a monoclonal antibody with a high affinity for VEGF A. The binding site is located at amino acid sites 88–89. Ranibizumab binds and inactivates all isoforms of VEGF, including the soluble VEGF isoforms 110, 121 and 165 and the tissue-bound isoforms 189 and 206.62 ,66 While bevacizumab was developed for long systemic retention in the treatment of metastatic cancer, ranibizumab was designed for rapid systemic clearance by removing the Fc fragment from the parent molecule.67 Additionally, the affinity of the compound for VEGF was enhanced by modification of five amino acids. In animal models, intravitreal injection effectively reduced retinal and choroidal neovascular growth as well as leakage from established vessels.68 Ranibizumab, with its molecular weight of 76 kDa, was found to penetrate the retina well after intravitreal injection.69 With a short intravitreal half-life time of 2–4 days, and a rapid systemic clearance, the systemic safety of ranibizumab is extremely high.70 Ranibizumab monotherapy has, therefore, become the reference standard for treatment of CNV.
Evidence
Fixed regimens were evaluated in the MARINA, ANCHOR, PIER and EXCITE studies. Seven hundred and sixteen patients with minimally classic or purely occult CNV, and evidence of presumed recent disease progression, were included in the MARINA study, a randomised, multicenter, sham-controlled phase 3 trial. Patients received monthly injections of 0.3 or 0.5 mg of ranibizumab or sham treatment continuously over 24 months. At 12 months, 95% of ranibizumab-treated eyes compared with 62% of sham-treated eyes, lost <15 letters in VA.13 Visual improvement by >15 letters was found in 34% of eyes treated with a dose of 0.5 mg (figure 10A and B). At 24 months, 90% of eyes in the 0.5 mg group had continued to maintain stable vision without loss of >15 letters compared with 53% in the control group.34 A mean improvement of seven letters was documented at 24-month follow-up. Thirty-three per cent of eyes in the 0.5 mg dose group improved by >15 letters with 42% ending up with a VA of 20/40 or better. Ranibizumab prevented further CNV growth and decreased the mean area of leakage angiographically in both dose groups. Typically, the functional and anatomical effects were seen rapidly within the first 3 months of intervention, and were maintained throughout the entire follow-up of 24 months.34 ,66 Additionally, patients treated with ranibizumab reported large improvements in near vision, distance vision and vision-specific dependency in quality-of-life questionnaires (NEI-VFQ-25).16
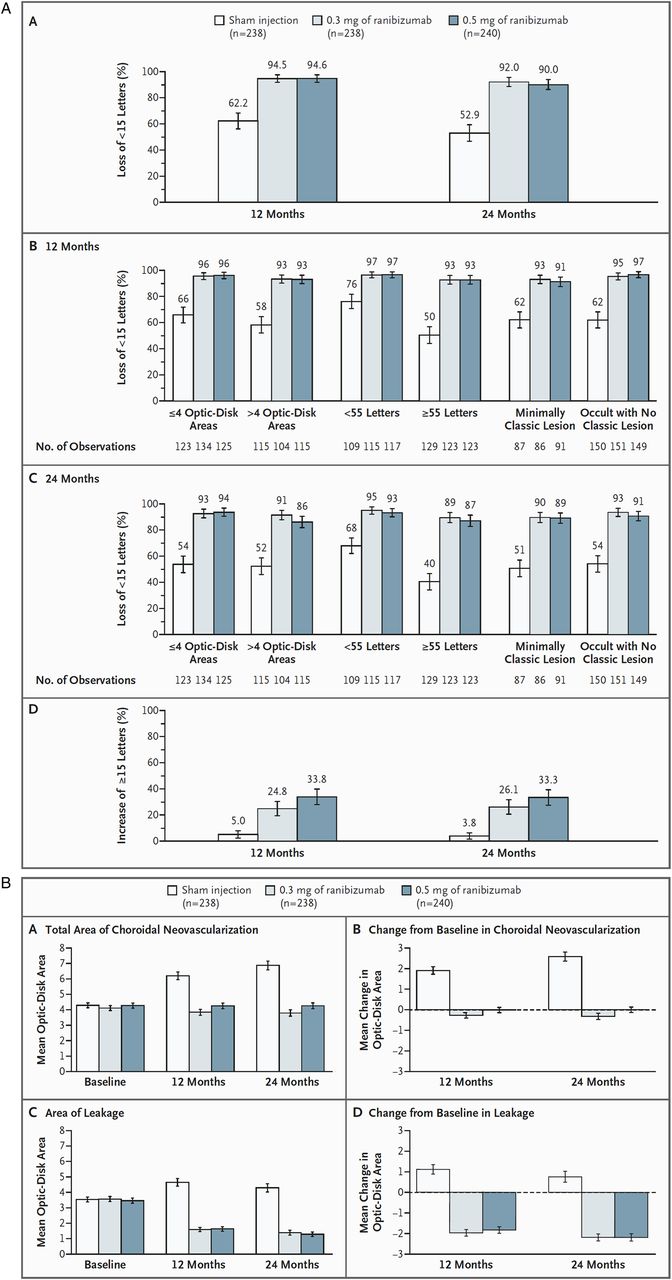
MARINA study. (A) Rate of loss or gain of visual acuity at 12 and 24 months associated with ranibizumab, as compared with sham injection. At 12 months, mean increases in visual acuity were +6.5 letters in the 0.3 mg group and +7.2 letters in the 0.5 mg group, as compared with a decrease of –10.4 letters in the sham-injection group (p<0.001 for both comparisons). The benefit in visual acuity was maintained at 24 months. The average benefit associated with ranibizumab over that of sham injection was approximately 17 letters in each dose group at 12 months, and 20–21 letters at 24 months. (B) Mean (±SE) changes in choroidal neovascularisation and leakage. The mean change from baseline in each of the ranibizumab-treated groups differed significantly from that in the sham-injection group at 12 and 24 months (p<0.001 for each comparison) in favour of ranibizumab treatment. Printed with permission from ref 13.
The ANCHOR study included 423 patients with predominantly classic subfoveal CNV due to AMD in a prospective, randomised phase 3 trial design.71 Monthly injections of ranibizumab at 0.3 or 0.5 mg were compared with standard PDT, which was indicated at 3-month intervals if leakage was seen angiographically. Ninety per cent of all eyes treated with ranibizumab at 0.5 mg lost less than 15 letters compared with 66% of eyes maintaining vision with PDT treatment alone at 24 months. Forty-one per cent of eyes in the 0.5ä-mg dose group improved by >15 letters and 12% improved by >30 letters, compared with 6% of PDT-treated eyes trated with PDT (figure 11). Additionally, these ranibizumab-treated patients demonstrated a mean improvement of 11 letters at 24 months, and 38% had a final outcome of 20/40 or better. Initial VA, or lesion size, had no impact on vision prognosis.66 ,72

ANCHOR study. Mean (±SE) changes in the number of letters read as a measure of visual acuity from baseline through 12 months. The tracking of mean changes in visual acuity scores over time showed that the values in each of the ranibizumab groups were significantly superior to those in the verteporfin group at each month during the first year (p<0.001) (figure 2) On average, visual acuity of ranibizumab-treated patients increased by +5.9 letters in the 0.3 mg group and +8.4 letters in the 0.5 mg group at 1 month after the first treatment and increased further over time to a gain of +8.5 letters in the 0.3 mg group and +11.3 letters in the 0.5 mg group by 12 months. By contrast, the verteporfin group had an average loss in visual acuity at each month after the first month, with a mean loss of 9.5 letters by 12 months. Printed with permission from ref 13.
The PIER study, a phase 3b trial, included 182 patients with all lesion types and evaluated the efficacy and safety of monthly ranibizumab at three doses followed by dosing every 3 months. While patients in the sham group lost a mean of 16 letters during 12 months of follow-up, patients receiving either dose of ranibizumab remained stable at baseline VA.66 Ninety per cent in the group receiving the 0.5 dose lost <15 letters compared with 49% in the sham group; 13% versus 10% gained >15 letters.73 As the overall VA, returned to baseline from month 3 to month 12 after switching to quarterly dosing, this reduction appears to suggest that quarterly dosing is inferior to monthly dosing. This was subsequently confirmed by the EXCITE study.50 Patients (n=353) with all types of CNV were randomised (1:1:1) to receive doses of ranibizumab at 0.3 mg quarterly, 0.5 mg quarterly or 0.3 mg monthly. Treatment comprised a loading phase (3 consecutive monthly injections) followed by a 9-month maintenance phase (either monthly or quarterly injection).50 Mean VA gain over baseline was observed for the entire 12-month trial in all groups. At month 12 compared with month 3, the VA gain was slightly decreased with quarterly dosing (by −2.2 and −3.1 letters with 0.3 mg and 0.5 mg of ranibizumab, respectively) (figure 12A –C).
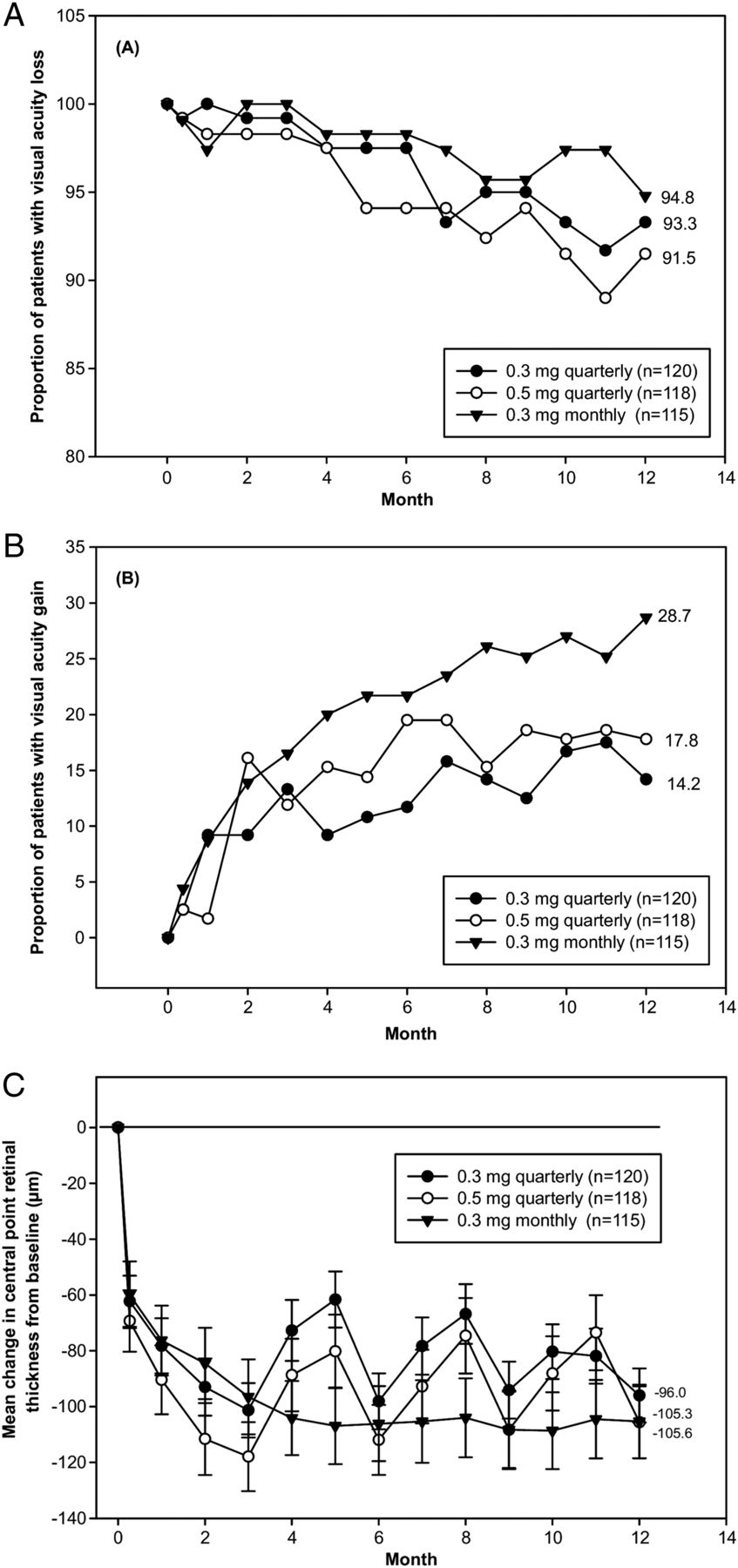
EXCITE study. (A, B) Proportion of patients with (A) visual acuity loss (<15 letters) or (B) gain (≥15 letters) over time in the intent-to-treat patient population (last observation carried forward (LOCF)) of EXCITE. Best-corrected visual acuity (BCVA) increased from baseline to month 12 by +4.9, +3.8, and +8.3 letters in the 0.3 mg quarterly (104 patients), 0.5 mg quarterly (88 patients), and 0.3 mg monthly (101 patients) dosing groups, respectively. After three initial monthly ranibizumab injections, monthly (0.3 mg) and quarterly (0.3 mg/0.5 mg) ranibizumab treatments maintained BCVA in patients with choroidal neovascularisation secondary to age-related macular degeneration. At month 12, BCVA gain in the monthly regimen was higher than that of the quarterly regimens. The non-inferiority of a quarterly regimen was not achieved with reference to 5.0 letters. (C) Mean change from baseline over time of central retinal thickness as assessed by optical coherence tomography scan in the intent-to-treat patient population ( LOCF) of EXCITE. Vertical bars represent SE of the mean. The mean decrease in central retinal thickness from baseline to month 12 in the intention-to-treat population was −96.0 µm in 0.3 mg quarterly, −105.6 µm in 0.5 mg quarterly, and −105.3 µm in 0.3 mg monthly group. Printed with permission from ref 50.
Flexible regimens were evaluated in the subsequent trials including PrONTO, CATT, SECURE and HARBOR. The small, open-label, prospective, non-randomised PrONTO study assessed three consecutive monthly injections followed by OCT-guided variable-interval dosing (at >1 month intervals).48 Retreatment criteria were a 5-letter loss and fluid as detected by OCT; >100 mm increase in CRT; new-onset classic CNV; new macular haemorrhage; or persistent macular fluid detected by OCT >100 mm increase in central thickness (CRT) new-onset classic CNV new macular haemorrhage or persistent macular fluid detected by OCT. Although VA outcomes were similar to those of the MARINA and ANCHOR trials with fewer intravitreal injections, substantial trial design differences limit comparisons. Despite small and open-label, this study suggested that flexible OCT-guided retreatment could sustain visual gain with fewer injections, a concept which has since become a popular model in clinical practice, particularly in Europe.
The investigators in the randomised CATT trials set out to assess the relative efficacy and safety of ranibizumab and bevacizumab and to determine whether an as-needed regimen, compared with a monthly regimen, would compromise long-term VA.54 The treatment protocol was much tighter than used previously, and is often referred to as ‘zero tolerance’. Radial scanning by TD-OCT was used in the trial and any fluid on OCT was added to the usual retreatment criteria. At 12 months, patients treated with monthly ranibizumab with 11.7 injections, and with ranibizumab as needed with 6.9 injections, gained +8.5 and +6.8 letters, respectively.54 Based on the trial design, PRN using ranibizumab was considered to be non-inferior. However, a meta-analysis combining the data from all the groups, as well as the data from the IVAN study, a similar trial in the UK with different retreatment protocol, found that discontinuous was inferior to continuous treatment.74 As the latter also included data from bevacizumab, the findings might have been different, if ranibizumab had been used alone, were in this analysis.
In the CATT year 2 data, when the monthly ranibizumab group was re-randomised into a continuous monthly treatment and as-needed treatment, the as-needed group lost −1.8 letters as compared with those staying with monthly treatment. Over the entire 2 years, the vision gain was very similar to the year 1 data with +8.8 and +6.7 letters in the monthly and as needed, respectively.54 In other words, changing to as needed in year 2 lost all the benefit of the monthly treatment from year 1.
SECURE75 and HORIZON76 are prospective extension studies that were designed to assess the long-term safety and efficacy of intravitreal injections of 0.5 mg of ranibizumab in patients with neovascular AMD. HORIZON is a multicentre, open-label, 24-month extension study following patients who had completed the MARINA, ANCHOR, or FOCUS (RhuFAb V2 Ocular Treatment Combining the Use of Visudyne to Evaluate Safety) trials. It is to evaluate long-term safety, tolerability and efficacy of multiple intravitreal injections of 0.5 mg ranibizumab to patients as needed, SECURE is a phase 4.2-year extension study in patients with AMD who had completed 1 year of treatment with ranibizumab in the EXCITE75 or SUSTAIN77 studies. The results from the SECURE study corroborate the findings from the HORIZON study,76 where there was an incremental decline in the BCVA gains achieved with monthly ranibizumab treatment in the previous studies, leading to an overall gradual decline in BCVA by −7.5 letters (ranibizumab-treated initial group) at the study end. This VA decline highlights the progressive nature of neovascular macular disease and shows a strict need for continuous follow-up monitoring and rigorous objective retreatment criteria. Continued follow-up in the SEVEN-UP study also suggested a long-term persistence of disease activity in the majority of patients.49
The extension studies have provided further data on the safety of ranibizumab treatment. Intravitreal injections of ranibizumab were associated with a low incidence of endophthalmitis (0.9%) in the SECURE study75 consistent with the rates reported in the HORIZON study (0.2% for presumed endophthalmitis).76 The rates are also consistent with those reported at 2 years in previous neovascular AMD studies (MARINA, 1.0%; ANCHOR, 1.1%). In the SECURE study, arterial thromboembolic events (ATEs) (categorised under adverse effects (AE) of special interest, and including haemorrhagic cerebrovascular conditions, ischaemic cerebrovascular conditions, myocardial infarction, and arterial embolic and thrombotic events) occurred in 5.6% of the patients receiving ranibizumab. These data are similar to the rate of ATEs (according to the Anti-platelet Trialists Collaboration criteria) reported in patients receiving ranibizumab in the ANCHOR and MARINA studies (4.4%–5%) and the HORIZON study (5.3% in the ranibizumab-treated initial patients).25
Additionally, the LUMINOUS programme was initiated as part of an ongoing pharmacovigilance programme for ranibizumab, it was designed to assess long-term safety, efficacy, treatment patterns, and health-related quality-of-life outcomes in a large number of patients treated with ranibizumab in routine clinical practice across the world.25 (ClinicalTrials.gov identifier: NCT01318941).
The HARBOR study is the only trial that has included SD-OCT monitoring into a PRN regimen compared with monthly treatment.55 The study evaluated the 12-month efficacy and safety of 0.5 and 2.0 mg intravitreal dosing of ranibizumab monthly and on an as-needed (PRN) basis in treatment-naive patients with subfoveal neovascular AMD. Patients (n=1098) were randomised to receive ranibizumab 0.5 or 2.0 mg ranibizumab injections intravitreally, monthly or on a PRN basis after three monthly loading doses. At month 12, the mean change from baseline in BCVA for the four groups was +10.1 letters (0.5 mg monthly), +8.2 letters (0.5 mg PRN), +9.2 letters (2.0 mg monthly), and +8.6 letters (2.0 mg PRN). The proportion of patients who gained ≥15 letters from baseline at month 12 in the four groups was 34.5%, 30.2%, 36.1% and 33.0%. The mean change from baseline in central foveal thickness at month 12 was −172, −161.2, −163.3 and −172.4 μm. The mean number of injections was 7.7 and 6.9 for the 0.5 mg PRN and 2.0 mg PRN groups. Ocular and systemic safety profiles were consistent with previous ranibizumab trials in AMD and similar between groups, without any safety risks documented. At month 12, the ranibizumab 2.0 mg monthly group did not meet the prespecified non-inferiority (NI) comparison. Vision in all treatment groups improved clinically meaningfully (+8.2 to +10.1 letters), and all groups had improved anatomic outcomes, with the PRN groups requiring approximately four fewer injections (6.9–7.7) than the monthly groups (11.2–11.3). (figure 13A and B). No new safety events were observed despite a fourfold dose escalation in the study. Therefore, the HARBOR study confirmed that 0.5 mg of ranibizumab dosed monthly provides optimum results in patients with neovascular AMD, and that there is no great disadvantage in using a PRN regimen instead of continued monthly injections55 provided that strict monthly monitoring is provided using SD-OCT technology.

HARBOR study. (A) Mean change from baseline to month 12 in best-corrected visual acuity (BCVA). *Vertical bars are ±1 SE of the unadjusted mean. Mean number of injections was analysed for patients who received at least 1 ranibizumab injection in the study eye. At month 12, the mean change from baseline in BCVA for the four groups was +10.1 letters (0.5 mg monthly), +8.2 letters (0.5 mg pro-re-nata (PRN)), +9.2 letters (2.0 mg monthly), and +8.6 letters (2.0 mg PRN). The proportion of patients who gained ≥15 letters from baseline at month 12 in the 4 groups was 34.5%, 30.2%, 36.1% and 33.0%, respectively. The mean number of injections was 7.7 and 6.9 for the 0.5 mg PRN and 2.0 mg PRN groups, respectively. (B) Mean change from baseline to month 12 in central foveal thickness (CFT) by spectral-domain optical coherence tomography. Vertical bars are ±1 SE of the unadjusted mean. The mean change from baseline in CFT at month 12 in the 4 groups was −172, −161.2, −163.3, and −172.4 μm, respectively. Printed with permission from ref 55.
Treat-and-extend is another flexible strategy suggested to reduce retreatment numbers. After three initial monthly ranibizumab or bevacizumab injections, and then to continue with monthly injections until there was no CNV activity (subretinal/intraretinal fluid, loss of >5 letters, or persistent/recurrent retinal haemorrhage) in a prospective cohort study of 120 patients.78 When there was no leakage activity, the interval to the next visit/injection was extended by 2 weeks to a maximum of 12 weeks. When there was CNV activity, this interval was shortened by 2 weeks. Mean VA change from baseline was +9.5±10.9 and +8.0±12.9 letters after 12 and 24 months, respectively, with, on average, 8.6±1.1 visits/injections in the first year, and 5.6±2.0 in the second year. After 12 and 24 months, 97.5% and 95.0% of patients, respectively, lost <15 letters. This ‘inject-and-extend’ protocol with fewer injections and visits delivered outcomes similar to those of the pivotal clinical trials of monthly ranibizumab with fewer injections and fewer visits. Treat-and-extend trials are currently underway in Europe. Although undertreatment is the major issue, complications from overtreatment should also be considered because, since a substantially increased rate of geographic atrophy (GA) was documented with monthly use of ranibizumab had new GA lesions after 2 years compared with only 15% of eyes treated in the as-needed arm.
Recommendation
Lucentis has been approved by the FDA since July 2006 for all lesion types in neovascular AMD in the USA since July 2006. An approval by the European Medicines Agency (EMA) for countries in the EU was granted in January 2007. The approved dose is 0.5 mg of ranibizumab. Giving injections continuously monthly for 2 years on a PRN regimen with strict monthly monitoring using SD-OCT and retreatment, whenever any evidence of fluid is noted by retinal imaging has been found to be the regimen that secures the optimum results in vision outcome. The official product label in Europe recommends monthly intravitreal injections continued until maximum VA is achieved for three consecutive monthly assessments. Thereafter, patients should be monitored monthly for VA. Treatment is to be resumed when monitoring indicates loss of VA due to wet AMD. Monthly injections should then continue until stable VA is reached again for three consecutive monthly assessments. Current usage in Europe is based on OCT monitoring and cessation of treatment when fluid is absent on OCT. Treat-and-extend is being evaluated in prospective clinical trials. Development of GA should be observed during prolonged treatment.
The recommendations are based on the ANCHOR, MARINA, PIER, EXCITE, HARBOR and CATT study data (evidence level I) as well as the SECURE and HORIZON study data (evidence level II).
Bevacizumab
Rationale
Bevacizumab is a full-length recombinant monoclonal antibody that binds all VEGF isoforms. It was developed to inhibit pathological angiogenesis in tumours and tumour growth and is approved by the FDA and EMA for the intravenous treatment of metastatic colorectal cancer and other cancer types.79 Cancer patients receiving systemic bevacizumab are commonly found to have an increased risk of cardiovascular events, stroke and gastrointestinal bleeding.80 A mathematical model comparing the time-dependent relative elimination of ranibizumab, bevacizumab and aflibercept was used to determine the theoretical peak and binding activities when the drugs were injected every 28 days. The intravitreal half-lives of ranibizumab, bevacizumab, and aflibercept were estimated to be 3.2, 5.6 and 4.8 days, respectively. The relative molar binding activities of ranibizumab, bevacizumab and aflibercept were 1, 0.05 to 0.2, and 140, respectively, indicating a lower binding affinity for bevacizumab.81 The systemic retention is prolonged because the Fc-portion of the substance binds to an endothelial cell receptor and is recycled. Intravitreal bevacizumab has recurrently been found to lower systemic VEGF concentrations much more than ranibizumab. Because bevacizumab and VEGF have similar bindings patterns, it is hypothesised that bevacizumab may be as effective as ranibizumab in the treatment of neovascular AMD and other types of intraocular neovascularisation, and may provide a less expensive alternative to approved substances specifically adapted for intraocular use.82–85
Evidence
Since 2005, many uncontrolled and retrospective case series have indicated that bevacizumab has a beneficial effect in the treatment of neovascular AMD.86–88 Bevacizumab has been split from the original vial into single doses containing 1.25 mg in a volume of 0.05 mL. Intraocular use has incidentally been associated with clusters of non-infectious mild to severe ocular inflammation (153 patients reported, no serious sequelae) and a single cluster of infectious endophthalmitis (12 patients), the latter associated with inappropriate pharmacy dispensing of the drug.89 ,90 A sterile preparation of single doses is mandatory with timely usage to prevent contamination spreading and aggregates forming, which leads to enhanced intraocular inflammatory reactions.91
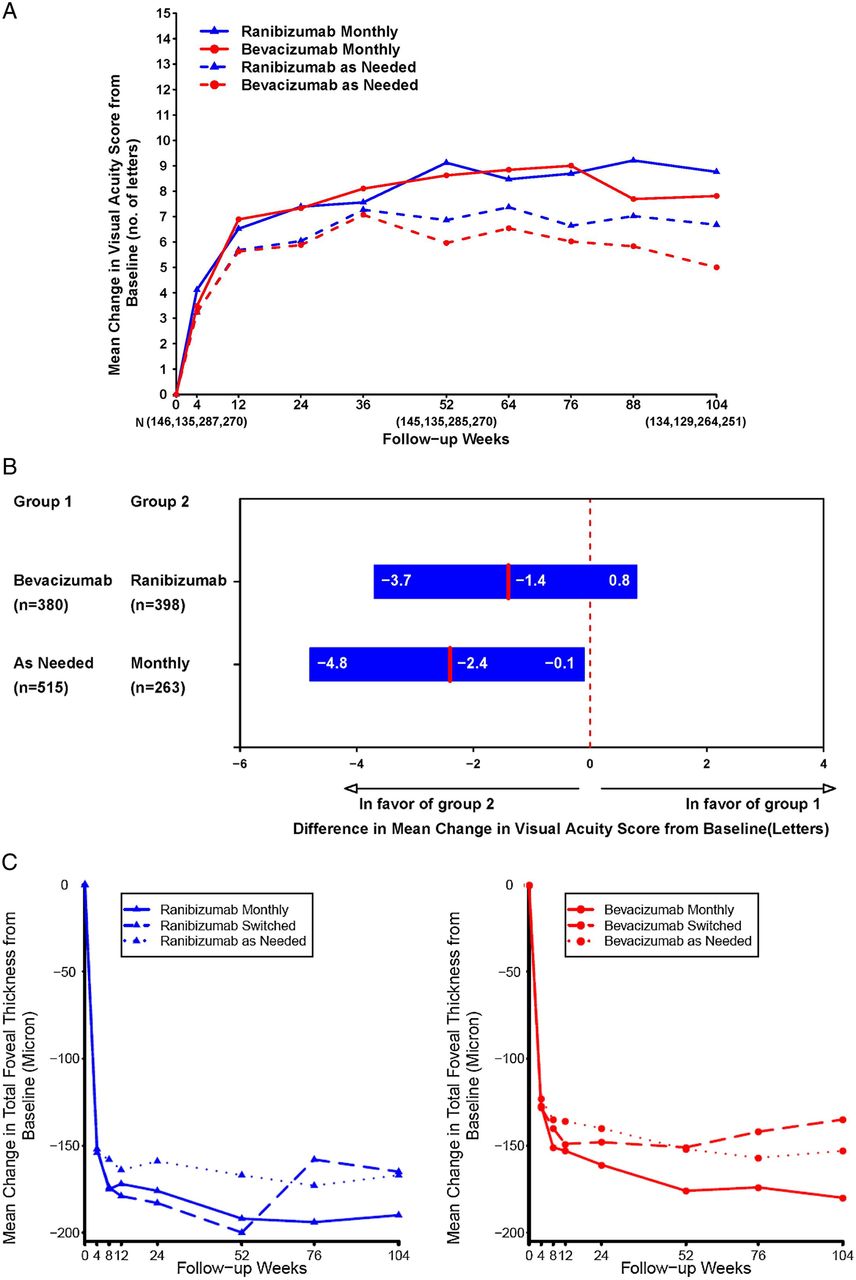
Evidence level I data for bevacizumab are exclusively derived from NI trials comparing bevacizumab with ranibizumab in the treatment of neovascular AMD aimed at reducing drug-related costs in clinical practice. The CATT study was a single-masked, NI trial, in which 1208 patients with neovascular AMD were randomised to intravitreal injections of ranibizumab or bevacizumab on either a monthly schedule or as needed, with monthly evaluation. At 1 year, with a difference of five letters, monthly bevacizumab was equivalent to monthly ranibizumab, with +8.0 and +8.5 letters gained.92 Bevacizumab as needed was equivalent to ranibizumab as needed with +5.9 and +6.8 letters gained. Nevertheless, the comparison between bevacizumab as needed and monthly bevacizumab was inconclusive, and NI was not achieved. The mean decrease in CRT was greater in the ranibizumab monthly group (196 μm) than in the other groups (152–168 μm). Rates of death, myocardial infarction, and stroke were similar for patients receiving either bevacizumab or ranibizumab (p>0.20). However, the proportion of patients with serious systemic adverse events (primarily hospitalisations) was higher with bevacizumab than with ranibizumab (24.1% vs 19.0%; risk ratio, 1.29; 95% CI 1.01 to 1.66). Subsequently, 1107 patients were followed during year 2, and the patients initially assigned to monthly treatment were reassigned randomly to monthly or as-needed treatment, without changing the drug assignment.54 The mean gain in VA was similar for both drugs (bevacizumab-ranibizumab difference, −1.4 letters; 95% CI), but greater for monthly than for as-needed treatment (difference, −2.4 letters; 95% CI −4.8 to −0.1; p=0.046). The proportion of eyes without fluid was 13.9% in the bevacizumab as-needed group, against 45.5% in the ranibizumab monthly group (drug, p=0.0003; regimen, p<0.0001 with statistically significantly more eyes treated with ranibizumab demonstrating resolution of fluid). Generally, switching from monthly to as-needed treatment resulted in a greater mean decrease in vision during year 2 (−2.2 letters; p=0.03) and a lower proportion without fluid (−19%; p<0.0001). Rates of death and arteriothrombotic events were similar for both drugs (p>0.60) after 2 years, but the proportion of patients with one or more systemic serious adverse events was again higher with bevacizumab than with ranibizumab (39.9% vs 31.7%; adjusted risk ratio, 1.30; 95% CI 1.07 to 1.57; p=0.009). Treatment as needed, generally resulted in less gain in VA, whether instituted at enrolment or after 1 year of monthly treatment. The differences between BCVA values at 2 years increased compared with the year 1 outcomes with monthly ranibizumab scoring highest and as-needed bevacizumab scoring lowest. Retreatment indications were based on loss in BCVA or morphologic evidence of fluid in the macula based on TD-OCT without clear definition of the type and localisation of fluid ‘no tolerance’ regimen, and no clear biomarkers were identified by the protocol (figure 14A-C).

CATT study. (A) The mean change in visual acuity from enrolment over time in patients treated with the same dosing regimen for 2 years. While ranibizumab monthly, becacizumab monthly and ranibizumab as needed meet the non-inferiority level, treatment with bevacizumab as needed led to inconclusive results and non-inferiority was not proven. At 2 years, the mean increase in letters in visual acuity from baseline was +8.8 in the ranibizumab monthly group, +7.8 in the bevacizumab monthly group, +6.7 in the ranibizumab as-needed group and +5.0 in the bevacizumab as-needed group. Main gain was greater for monthly than for as-needed treatment. Switching from monthly to as-needed treatment resulted in greater mean decrease in vision during year 2 with −2.2 letters. (B) Differences in mean change in visual acuity at 2 years and 95% CIs in patients treated with the same dosing regimen for 2 years. The difference in mean improvements for patients treated with bevacizumab relative to those treated with ranibizumab was −1.4 letters. The difference in mean improvements for patients treated by an as-needed regimen relative to those treated monthly was −2.4 letters. (C) The mean change in total foveal thickness from enrolment over time by dosing regimen within drug group: (A) ranibizumab and (B) bevacizumab. Mean gain was greater for monthly than for as-needed treatment. The proportion without fluid ranged from 13.9% in the bevacizumab as-needed group to 45.5% in the ranibizumab monthly group. Printed with permission from ref 54.
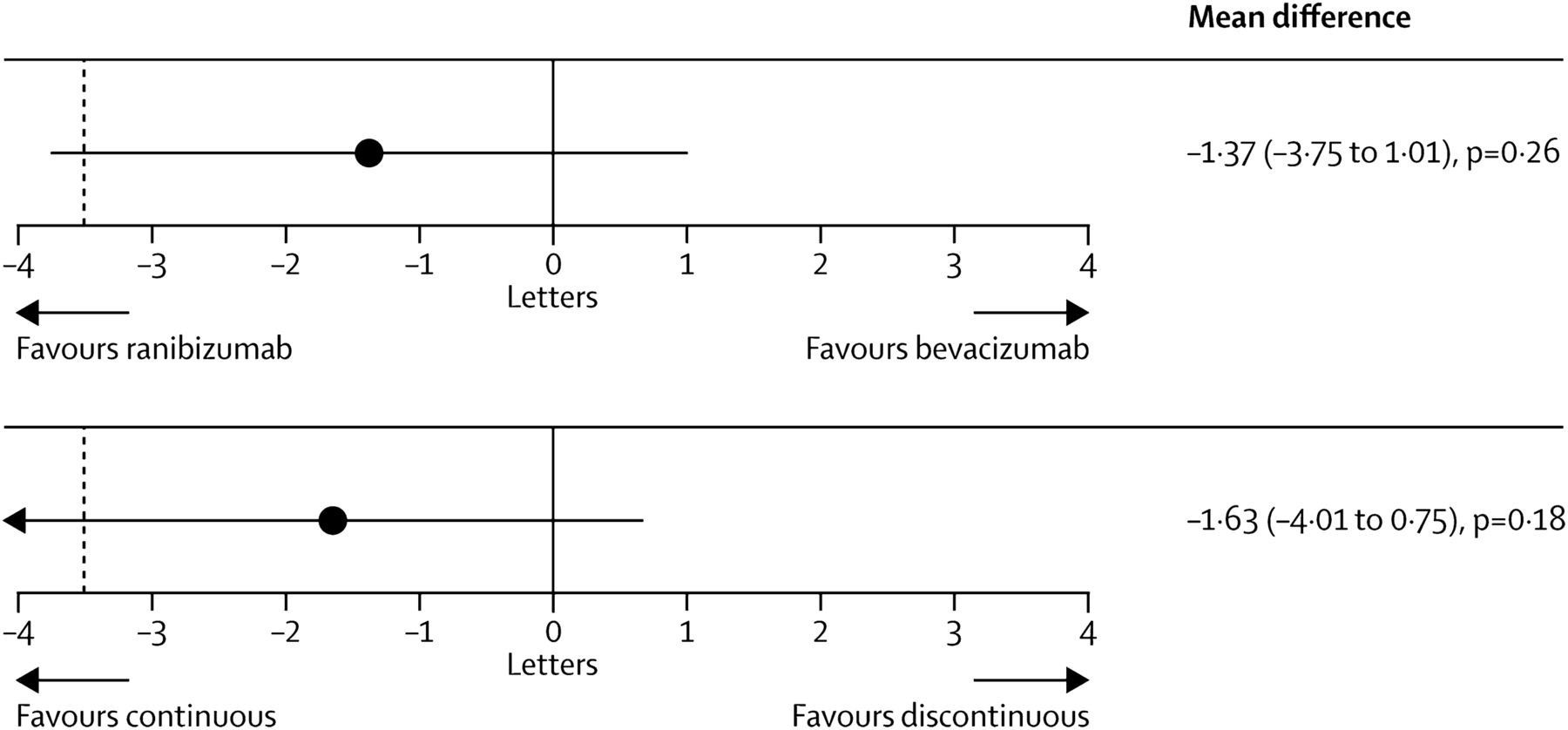
In the IVAN study, 610 patients were assigned randomly to ranibizumab or bevacizumab, given either every month (continuous) or as needed (discontinuous), with three consecutive injection series and monthly review. One year after randomisation, the comparison between bevacizumab and ranibizumab was inconclusive and bevacizumab did not meet the NI criteria (bevacizumab minus ranibizumab −1.99 letters, 95% CI −4.04 to 0.06)74. Discontinuous treatment was equivalent to continuous treatment (discontinuous minus continuous −0.35 letters; 95% CI −2.40 to 1.70). There was no difference between drugs in the proportion of patients experiencing a serious systemic adverse event (OR, 1.35; 95% CI 0.80 to 2.27; p=0.25). Serum VEGF was lower with bevacizumab (geometric mean ratio, 0.47; 95% CI 0.41 to 0.54; p<0.0001) and higher with discontinuous treatment (geometric mean ratio, 1.23; 95% CI 1.07 to 1.42; p=0.004). After 2 years, bevacizumab similarly failed to fall within the NI margin (mean difference −1.37 letters, 95% CI −3.75 to 1.01; p=0.26).93 Moreover, discontinuous treatment also did not reach the NI level (−1.63 letters, −4.01 to 0.75; p=0.18), that is, the reduction in the frequency of retreatment resulted in a small loss of efficacy irrespective of the chosen drug. Frequency of arterial thrombotic events or hospital admission for heart failure did not differ between groups given ranibizumab (20 (6%) of 314 participants) and bevacizumab (12 (4%) of 296; OR 1.69, 95% CI 0.80 to 3.57; p=0.16), or those given continuous (12 (4%) of 308) and discontinuous treatment (20 (7%) of 302; 0.56, 0.27 to 1.19; p=0.13). Mortality was lower with continuous than discontinuous treatment (OR 0.47, 95% CI 0.22 to 1.03; p=0.05), but did not differ by drug group (0.96, 0.46 to 2.02; p=0.91).
With respect to safety, pooled analyses of the CATT and IVAN studies showed that mortality was lower with ranibizumab, but neither outcome differed significantly between drugs with the size of the respective study population (p=−0.34 and p=−0.55). Increased odds of experiencing a serious adverse event with bevacizumab observed in the CATT persisted in the meta-analysis (p=−0.016). Most importantly, the CATT and IVAN studies were not powered to identify small, but clinically significant differences in the safety of the two compounds (figure 15).
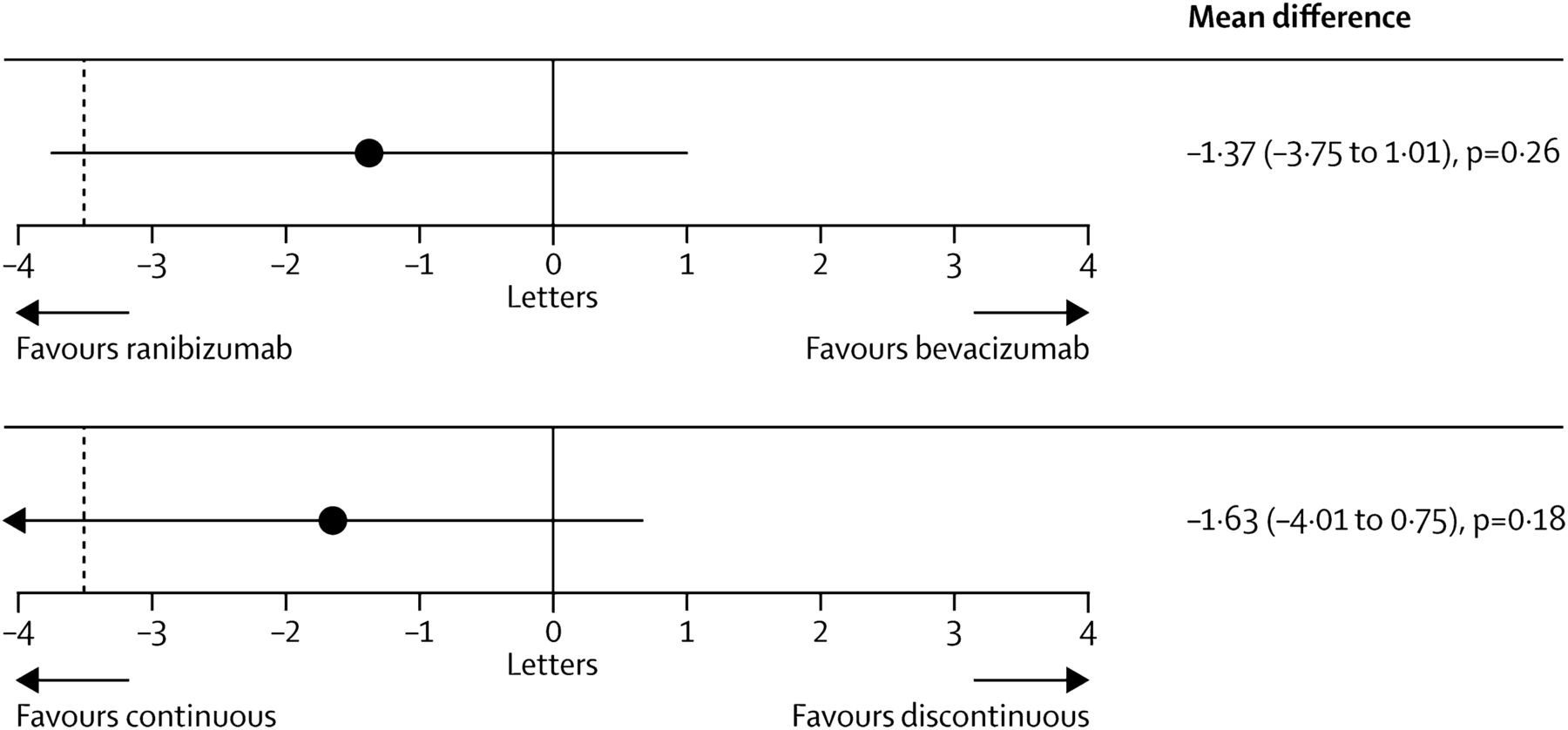
IVAN study. Mean differences in best corrected distance visual acuity at 2 years by drug (top) and by regimen (bottom). Black dashed line shows non-inferiority limit of −3.5 letters. Mean differences estimated with data from visits 0, 3, 6, 9, 12, 15, 18, 21, and 24, adjusted for centre size. For best-corrected visual acuity, bevacizumab was neither non-inferior nor inferior to ranibizumab (mean difference −1.37 letters, 95% CI −3.75 to 1.01; p=0.26). Discontinuous treatment was neither non-inferior nor inferior to continuous treatment (−1.63 letters, −4.01 to 0.75; p=0.18). Printed with permission from ref 93.
The GEFAL study was a multicenter, prospective, NI, double-masked, randomised, clinical trial performed at 38 French ophthalmology centres.94 Patients were randomly assigned to receive intravitreal bevacizumab (1.25 mg) or ranibizumab (0.50 mg). Hospital pharmacies were responsible for preparing, blinding and dispensing treatments. Patients were followed for 1 year, with a loading dose of three monthly intravitreal injections, followed by an as-needed regimen (1 injection in the case of active disease) for the remaining 9 months with monthly follow-up. Five hundred and one patients were assigned randomly. In the per protocol analysis, bevacizumab was non-inferior to ranibizumab (bevacizumab minus ranibizumab +1.89 letters; 95% CI −1.16 to +4.93, p<0.0001). The mean number of injections was 6.8 in the bevacizumab group and 6.5 in the ranibizumab group (p=0.39). Both drugs reduced the central subfield macular thickness, with a mean decrease of 95 μm for bevacizumab and 107 μm for ranibizumab (p=0.27). There were no statistically significant differences in the presence of subretinal or intraretinal fluid at final evaluation, dye leakage on angiogram or change in choroidal neovascular area, but ranibizumab tended to have a better anatomic outcome. The proportion of patients with serious adverse events was 12.6% in the bevacizumab group and 12.1% in the ranibizumab group (p=0.88). The proportion of patients with serious systemic or ocular adverse events was similar in both groups.
Safety is a topic of controversy discussed issue in the use of bevacizumab. Experimental studies revealed that systemic VEGF inhibition disrupts endothelial homeostasis and accelerates atherogenesis, suggesting that these events contribute to the clinical cardiovascular adverse events of VEGF-inhibiting therapies. The recommendation was, therefore, to determine cardiovascular safety profiles to improve patient selection for therapy and allow close monitoring of patients at increased cardiovascular risk.95 In human studies, Avery et al96 found that the systemic exposure after the third monthly intravitreal injection was 13-fold greater for aflibercept and 70-fold greater for bevacizumab than for ranibizumab. Another report reviewed differences in both ocular and systemic safety between intravitreal bevacizumab and ranibizumab in the setting of neovascular AMD.91 Serious adverse events associated with either bevacizumab or ranibizumab injections are generally rare. Acute intraocular inflammation tends to occur more frequently following bevacizumab injection. Systemic absorption of bevacizumab is greater than with ranibizumab, and many studies have shown that specific risk or age groups of patients have an increased risk of systemic adverse events when receiving bevacizumab compared with those receiving ranibizumab. A systemic review based on Medline, Embase and the Cochrane Library evaluated whether bevacizumab is as safe as ranibizumab, and whether bevacizumab can be justifiably offered to patients as a treatment for AMD with robust evidence of no differential risk.97 Registered clinical trials that investigated bevacizumab or ranibizumab in direct comparison, or against any other control group (indirect comparison), and had a minimum follow-up of 1 year were included. Direct comparison (3 trials, 1333 patients): The 1-year data show a significantly higher rate of ocular AE with bevacizumab than with ranibizumab (relative risk (RR)=2.8; 95% CI 1.2 to 6.5). The proportion of patients with serious infections and gastrointestinal disorders was also higher with bevacizumab than with ranibizumab (RR=1.3; 95% CI 1.0 to 1.7). Arterial thromboembolic events were equally distributed among the groups. Indirect comparison: Ranibizumab versus any control (5 trials, 4054 patients). The 2-year results of three landmark trials showed that while absolute rates of serious ocular AE were low (≤2.1%), relative harm was significantly raised (RR=3.1; 95% CI 1.1 to 8.9). Bevacizumab versus any control (three trials, 244 patients): the safety profile of bevacizumab could not be judged due to the poor quality of AE monitoring and reporting in the trials. In summary, evidence from head-to-head trials raised concern about an increased risk of ocular and systemic adverse events with bevacizumab. The need for studies that are powered not just for efficacy, but also for defined safety outcomes based on the signals detected in systematic reviews must be emphasised.
Recommendation
The CATT and IVAN results indicate that ranibizumab and bevacizumab both confer solid visual function benefits. With monthly use of both drugs, NI has been proven with optimal visual outcomes. Direct comparison among as-needed treatments also demonstrated NI, although on a generally lower level. Bevacizumab, as needed, failed to meet NI equivalence to monthly ranibizumab, that is, bevacizumab used in a PRN regimen did not reach the superior visual outcome achievable with monthly ranibizumab. Therefore, choice of the proper (fixed monthly) regimen is relevant when off-label bevacizumab is used. How much reduction in ocular efficacy one would be willing to sacrifice for reducing the number of injections and/or costs might depend on individual circumstances.
No major safety issues have emerged, but conclusive data are lacking, and none of the trials were powered for safety. Nevertheless, evidence from head-to-head trials consistently raises concerns about an increased risk of ocular and systemic adverse events with bevacizumab. Bevacizumab's impact on plasma concentrations of VEGF and its prolonged half-life in the circulation are proven. Therefore, the individual physical condition of each patient should be considered in the choice of the adequate therapy with the notion that patients included in clinical trials do not reflect the common risk profile of the real-world population. Bevacizumab is substantially less expensive, but each treatment decision is—legally and medically—based on an individual agreement between treating physician and patient, and must be the consequence of a comprehensive discussion of treatment alternatives and incalculable risks. Informed consent after discussing the optimal benefit, comfort and risks and the off-label status of the drug is mandatory.98 Currently, there is a conflict on Avastin between the cost-conscious health authorities in EU member states and the EU drug regulators. There are several examples of cost-cutting solutions by health authorities, which risk undermining the fundamental principles of the regulatory framework. Meanwhile patients and doctors shoulder the risk.
These recommendations are based on the CATT and IVAN data (evidence level I).
Aflibercept
Rationale
Aflibercept, unlike the monoclonal antibodies, bevacizumab and ranibizumab, is a soluble decoy receptor fusion protein. In aflibercept, the second binding domain of the native VEGF receptor 1 and the third binding domain of VEGF receptor 2 are attached to the Fc component of human ICG.99 Therefore, the binding affinity of aflibercept (KDa=0.49 pmol/L) is higher than that of ranibizumab (KDa=0.46 pmol/L) and bevacizumab (KDa=0.58 pmol/L).100 ,101 The molecular size of aflibercept of 115 kDa results in an intravitreal half-life of 7.1 days, a calculated bioactivity in human eyes of 2.5 months and a serum half-life of 18 days due to the presence of an Fc portion.100 ,101 The compound binds to all VEGF-A isoforms and VEGF-B, with higher affinity than their native receptors.102 Aflibercept and ranibizumab have been found to be equally effective in blocking endothelial cell proliferation, and 10-fold more potent than bevacizumab.103 Aflibercept also binds to placental growth factor (PlGF) present on endothelial cells and leucocytes.100 In preclinical studies, the compound suppressed CNV and VEGF-induced vascular breakdown in mice and rats.104 ,105 However, prolonged and high-doses administration led to loss of endothelial cells and pericytes.106 Intravenous therapy in the first clinical trial was associated with systemic toxicity, such as proteinuria and hypertension.107
Evidence
The first intravitreal phase I study showed that aflibercept decreased macular oedema and SRF for at least six weeks and was well tolerated.108 A subsequent phase 2 clinical trial, CLEAR-IT2, compared monthly with quarterly intravitreal aflibercept at 0.5 and 2.0 mg. With both doses, monthly treatment was functionally and anatomically more efficient than quarterly treatment, initially and after a PRN regimen, until week 52.109 Approval of aflibercept for the treatment of neovascular AMD was based on two parallel phase 3 pivotal trials, VIEW 1 and VIEW 2.110 ,111 Two thousand four hundred and nineteen patients with treatment-naive neovascular AMD were included in these double-masked, multicentre, parallel-group, active-controlled, randomised trials. Eyes with new-onset subfoveal or juxtafoveal CNV comprising at least 50% of the total lesion size and a BCVA level between 20/40 and 20/320 Snellen equivalents were randomised 1:1:1:1 to two doses (0.5 and 2.0 mg) and two regimen (monthly and eight-weekly with 2.0 mg) of aflibercept. There was one control arm receiving ranibizumab (0.5 mg) at monthly intervals for 52 weeks. The primary endpoint was defined as NI in the proportion of patients maintaining BCVA. Secondary outcomes were change in BCVA and a reduction in CRT on OCT. At 52 weeks, all aflibercept groups, independent of doses and regimen, were non-inferior to the control group with equal maintenance of vision in 95% of eyes.110 A mean improvement of +8.7 letters (0.5 mg/q4 ranibizumab) in the control group compared with a mean change in BCVA of +9.3 letters with 2 mg aflibercept every 4 weeks and +8.4 letters every 8 weeks. In the integrated analysis of VIEW 1 (US centres) and VIEW 2 (centres in Canada, South America, Europe, Asia, Australia) all regimens were within 0.5 letters of the reference arm ranibizumab. Notably, in the VIEW 1 study alone, which included 1217 patients, the four weekly aflibercept regimen provided a benefit that was statistically superior to that seen in the other groups, with a visual gain of +10.9 letters.110 Lesions in the VIEW 1 trial were primarily smaller and associated with higher initial BCVA scores. Anatomically, all treatment groups demonstrated a similar rapid decline in CRT by −130 to −157 µm. In the eight-weekly aflibercept groups, bimonthly fluctuations in CRT were seen with recurrent exudation between extended aflibercept injections. Ocular and systemic adverse events were similar across all treatment groups with no statistically significant differences in Anti-Platelet Trialists' Collaboration (APTC) ATE events or AE rates.
A capped PRN regimen was applied to the 2457 patients in the complete trial from week 52 to week 96.111 Criteria for retreatments were new or persistent fluid on OCT, an increase in CRT of 100 µm or more, or loss of five Early Treatment Diabetic Retinopathy Study (ETDRS) letters or more, compared with the best previous score with fluid on OCT, new classic CNV seen by FA or haemorrhage on ophthalmoscope, and a mandatory ‘capped’ injections at an interval of 12 weeks since the previous treatment. The proportion of maintenance in BCVA ranged between 91% and 92% at week 96 for all groups. Mean BCVA gains were between +6.6 (aflibercept 0.5 mg) and +7.9 letters (ranibizumab q4), 7.6 letters (aflibercept q4 and q8) confirming NI for aflibercept and the eight-weekly retreatment regimen. (figure 16A and B). Overall, a mean loss of 0.8–1.7 letters was seen in all groups after the switch from a fixed to a capped flexible regimen. The retreatment frequency was similar for both substances during the capped PRN year, with 4.1/4.2 (aflibercept 2q4 and 2q8) and 4.7 (ranibizumab) injections. Less patients with pronounced disease activity requiring at least six reinjections during the second year received aflibercept (2 mg) (14%–16%) than ranibizumab (26.5%) treatments. Accordingly, more aflibercept-treated than ranibizumab-treated eyes were seen without retinal fluid at weeks 52 and 96.111

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
VIEW studies. (A) Mean change from baseline in best-corrected visual acuity (BCVA). The inset shows the difference in least square mean (with 95% CI) between intravitreal aflibercept arms and ranibizumab (aflibercept minus ranibizumab) for BCVA change from baseline to week 96, full analysis set. Outcomes for the aflibercept and ranibizumab groups were similar at weeks 52 and 96. Mean BCVA gains were 8.3–9.3 letters at week 52 and 6.6–7.9 letters at week 96. Patients received, on average, 16.5, 16.0, 16.2 and 11.2 injections over 96 weeks and 4.7, 4.1, 4.6 and 4.2 injections during weeks 52 through 96 in the Rq4, 2q4, 0.5q4, and 2q8 groups, respectively. All aflibercept and ranibizumab groups were equally effective in improving BCVA and preventing BCVA loss at 96 weeks. (B) Mean change from baseline central retinal thickness, full analysis set. Bimonthly fluctuations in central retinal thickness (CRT) are seen during the fixed regimen in year 1 in the 2q8 arm. During the second year with a capped pro-re-nata regimen, variations in CRT become larger with a quarterly fluctuation pattern. Printed with permission from ref 111.
Recent subgroup analyses of the VIEW trials suggested a superior morphologic efficiency of aflibercept in reducing intraretinal fluid and SRF as well as reducing RPE elevation. One retrospective assessment of eyes with persistent subfoveal fluid, despite previous treatments with ranibizumab suggested that at 6 months, mean BCVA and CRT improved significantly upon a switch to intravitreal aflibercept.112 However, most case series have indicated that 2.0 mg aflibercept leads to anatomic improvements in patients with long-standing persistent fluid, who have received other anti-VEGF agents, but BCVA conditions usually remained unchanged.113 ,114 Most of these analyses were retrospective in design and did not specify the previous anti-VEGF regimen and continuity in detail.
Recommendation
FDA approval of aflibercept for the treatment of neovascular AMD at a recommended intravitreal dose of 2.0 mg was granted in 2012. The suggested regimen is monthly injections for the initial 3 months followed by a fixed dosing every eight weeks. The label also highlights that no additional efficacy was demonstrated when aflibercept was dosed every 4 weeks compared with every 8 weeks. In the VIEW 1 study, however, monthly aflibercept provided statistically superior visual gains. Many interventional studies suggest a superior anatomic efficacy of aflibercept compared with ranibizumab and bevacizumab.
The EU label recommends three initial injections at monthly intervals, followed by eight weekly injections without any subsequent monitoring. After 12 months of treatment, the injection intervals may be prolonged, depending on the functional and anatomical condition of the individual patient. Control intervals for evaluation may be adjusted at the treating ophthalmologist's discretion. The bimonthly fluctuations in BCVA and CRT values are small when averaged but are more impressive individually. This suggests patients with more intensive desease activity who can benefit from a monthly regimen, as noted in the VIEW 1 study, should be identified. Beyond the first year, increased dosing with continued monthly/bimonthly injections may be needed in eyes with morphologic signals, such as PED and IRC, which are likely associated with aggressive progression of CNV disease. Evidence level I is provided by the VIEW I and II studies.110 ,111
Photocoagulation and PDP for neovascular AMD
Rationale
Photocoagulation relies on the high transparency of the neurosensory retina and the melanin pigment content of the RPE to exert a selective effect on the outer layers of the retina. Such photocoagulation can be used to obtain immediate closure of subretinal neovascular membranes (CNV) resulting in permanent cessation of exudation, haemorrhage and vessel growth. An alternative method of closing subretinal CNV is PDT, which combines intravenous infusion of a photosensitive dye that releases free oxygen radicals when exposed to targeted illumination of the area of the fundus where the new vessels are located. Activation of the photosensitiser occurs at light intensities below the threshold for thermal coagulation, and PDT, therefore, leads to less collateral tissue damage than photocoagulation. Both treatment modalities have been tested at highly powered levels in AMD associated with subfoveal CNV under the foveal centre or close to the fovea.
Photocoagulation therapy
Evidence
Photocoagulation therapy for neovascular AMD was developed gradually and mostly on a case-to-case basis, until it was tested on a large scale against sham photocoagulation in the Macular Photocoagulation Study (MPS) in the USA115 ,116 and in randomised studies in the UK117 and France.118 These studies were conducted in an era, when photocoagulation was the only effective form of treatment for neovascular AMD. In the MPS, the long-term visual prognosis for extrafoveal CNV lesions that were well delineated on fluorescein angiograms, and no parts of which were closer than 200 µm from the centre of the foveal avascular zone, was significantly improved by prompt intense and confluent photocoagulation covering the entire CNV. After 5 years, severe visual loss (30 ETDRS letters or more) occurred in 46% of treated eyes, as opposed to 64% of untreated eyes.119 Recurrence of CNV caused most of the visual loss between the early post-treatment period and 5 years after photocoagulation in the treatment group. A subsequent MPS examined the effect of photocoagulation of CNV lesions that extended to within 1–199 µm of the centre of the foveal avascular zone. After 1 year, severe visual loss had occurred in 31% of treated eyes and in 45% of untreated eyes. After 5 years, the respective proportions were 54% and 57%, persistent or recurrent CNV activity being responsible for most of the additional loss.120
Finally, an MPS of laser photocoagulation for subfoveal CNV extending under the centre of the foveal avascular zone showed that eyes that had not undergone prior photocoagulation suffered an immediate loss of vision after treatment, but gained a relative long-term benefit.121 After 24 months, the proportion of patients who had suffered such severe visual loss was still only 20% in the treated group, but it was 37% in the untreated group. The results apply only to CNV with a classic component, a well-defined margin on FA, and the greatest linear diameter of less than 3.5 disk diameters. Green argon laser (514 nm) was eventually considered state-of-the-art for photocoagulation of CNV because the summary experience of trying laser light of other colours and absorption characteristics is that the fundamental effect on CNV and the adjacent tissue components is the same. Green 532-nm frequency-doubled Nd-YAG laser have replaced argon laser and are now the standard in photocoagulation for CNV because they are more compact, and spectral shift has little or no effect on the tissue, these are now the standard in photocoagulation for CNV.
Photodynamic therapy
The principle of PDT of CNV in AMD has led to the clinical development and marketing of a single pharmaceutical product (Visudyne, Novartis Pharma AG, Basel, Switzerland) with the active ingredient verteporfin. Semi-selective accumulation of verteporfin in proliferating endothelial cells and subsequent activation of the drug by 689 nm light in the part of the fundus where the CNV is found, enables closing of the new vessels without damage to the overlying neurosensory retina or its blood vessels. The vessels of the underlying normal choroidal vessels are closed to varying degrees, but the choroidal vessels recover and become perfused again within a few weeks after treatment. This temporary closure does not in itself appear to cause any immediate damage to the retina. The long-term morphological response of the CNV is variable, from flattening of the retina and lasting quiescence after a single treatment, to recurrence of CNV activity within a few months, and disease progression despite several retreatments. Years after initiation of PDT for neovascular AMD outer retinal atrophy and scarring is often seen in the area of the CNV where PDT was administered.
Evidence that PDT is of functional benefit to patients with neovascular AMD stems from two parallel, identically designed randomised controlled double-masked 2-year clinical trials, which were reported together in the peer-reviewed literature and are known as the TAP Study.122 The study showed that PDT, administered promptly at baseline and optionally, depending on CNV activity, again every 3 months, was safe and effective in that it reduced the risk of moderate (15 ETDRS letters) and severe (30 ETDRS letters) loss of BCVA. The outcome was most favourable in eyes with predominantly classic (type 2) CNV: 59% of treated eyes lost less than 15 letters over 2 years versus 31% in the sham group. The number of treatments given was 3.4 in the first year and 2.2 in the second year. A phase 3 randomised, controlled, double-masked, 2-year trial known as the VIP trial was conducted in eyes with occult (type 1) CNV and no classic component or evidence of recent disease progression. A phase 3 randomised, controlled, double-masked 2-year trial known as the VIP trial was conducted.123 A retrospective subgroup analysis showed benefit of PDT in eyes with lesions smaller than four MPS disk areas or VA worse than 20/50, where treatment was associated with loss of 15 or more ETDRS letters in 25% of eyes compared with 51% of eyes in the sham treatment group. A subsequent trial of PDT in occult-only CNV failed to confirm a treatment benefit of PDT.124 Acute severe VA loss was the only clinically significant adverse event observed in PDT studies in AMD with CNV, such acute loss of more than 20 ETDRS letters occurring in less than 5% of the patients. Two randomised trials of PDT combined with ranibizumab failed to show a benefit of combination therapy over ranibizumab monotherapy.125 ,126 A recent retrospective study suggests that rescue therapy with the combination of an intravitreal anti-VEGF agent and PDT may benefit eyes that have failed anti-VEGF monotherapy by improving vision, eliminating fluid and reducing the need for anti-VEGF retreatment.127
Recommendation
Laser photocoagulation therapy and verteporfin PDP have shown benefits compared with the natural course in selected subtypes and stages of neovascular AMD. While largely superseded by intravitreal pharmaceutical VEGF inhibition, these two older forms of CNV treatment remain a rational therapeutic option for selected patients in whom VEGF inhibition is not advisable. Although the evidence is based on studies of wet AMD with neovascularisation under or very near the foveal centre, application of photocoagulation or PDT for subretinal new vessels is likely to be considered in current clinical practice in less common conditions, such as peripapillary CNV and, in the case of photocoagulation, in conditions such as extrafoveal CNV in pregnant women in whom neither PDT nor intravitreal VEGF inhibitors have been shown to be safe. Evidence I is provided by MPS, TAP and VIP studies.
Radiation therapy
Rationale
Radiation therapy, as monotherapy or combined with anti-VEGF treatment, has also been studied as a new option for the treatment of neovascular AMD. Although anti-VEGF treatments provide patients with impressive benefits in terms of VA, all require regular intravitreal injections and they generally suppress rather than eliminate the disease activity. As replicating tissue is more susceptible to the effects of radiation than non-replicating tissue, choroidal neovascular lesions, which consist of proliferating endothelial cells, are more sensitive to radiation treatment than normal non-proliferating capillary endothelial cells and larger vessels.128 The role of radiation therapy for neovascular AMD was explored as early as 1993, and the rationale for this approach was based on the known effects of radiation therapy on tumour microvasculature. Radiation treatment is able to prevent proliferation of vascular tissue by its anti-inflammatory, antiangiogenic and antifibrotic properties.129 ,130 Radiation used for medical therapies can be divided into two main categories depending on its method of delivery to the tissue. Brachytherapy uses a radiation source delivered directly to the lesion by surgery. The source is usually an isotope which produces ionising radiation as it decays and emits energy. Teletherapy (external beam therapy) uses radiation formed into a beam which can be projected at an internal body tissue from an external source. The source can also be an isotope, but more recently electronically produced ionising radiation has been used.131
Evidence
Initial studies investigated external beam radiation to treat neovascular AMD. While some studies showed results better than the natural history, the results do not compare favourably with those in the anti-VEGF era. This may be because of collateral damage to ocular tissue, and the difficulty of targeting macular lesions with a technology that was designed for larger lesions, such as tumours. Another factor could be the time delay before radiation has an effect. There is historical evidence that treatment with external beam radiation therapy and plaque brachytherapy for AMD can prevent progression of CNV membranes. However, the VA results have been less impressive, which is likely due to inaccurate targeting in the case of external beam radiotherapy or difficulty in optimising the seed placement in plaque brachytherapy.132
Currently, two different approaches to radiation therapy are being investigated: epimacular brachytherapy (EMBT) (VIDION) and stereotactic radiosurgery (IRay system).131 Radiotherapy produces a delayed response, but has a much longer duration of action. Therefore, there is a good scientific rationale for a synergistic response. Anti-VEGF therapy inhibits growth factors in the local area, while radiotherapy inhibits the local inflammatory cell population and induces an apoptotic effect on vascular endothelium. The overall result from these two approaches has the potential to offer a faster and more complete recovery of functional vision.
EMBT is designed as a system to deliver intraocular radiation by placing the source of radiation close to the CNV complex at the macula. This device uses β radiation from a strontium-90/yttrium-90 source, and as β radiation has a rapid decline in dose with increasing distance from the source, it limits the radiation exposure to a defined region with little damage to adjacent tissues. The β radiation used in epimacular brachytherapy is delivered via pars plana vitrectomy (PPV) surgery. Once the vitreous has been removed, the surgeon positions the probe over the CNV lesion. The probe is held in position for approximately 4 min and then removed. Delivery of radiation to neighbouring structures is low with this method. The macular lesion, the optic nerve and the lens receive, respectively, 24 grey (Gy), 2.4 Gy and 0.00056 Gy.131
The MERITAGE study was a prospective, multicentric interventional, non-controlled, clinical trial designed to evaluate the safety and efficacy of EMBT for the treatment of chronic, active neovascular AMD. Fifty-three eyes of 53 participants with CNV requiring frequent anti-VEGF retreatments were included. Participants underwent PPV with a single 24-Gy dose of EMB delivered by the method described above. Participants were retreated with ranibizumab monthly as needed (PRN), using predefined retreatment criteria. Before enrolment, participants received an average of 12.5 anti-VEGF injections. After a single treatment with EMB, 81% maintained stable vision, with a mean of 3.49 anti-VEGF retreatments in 12 months. Mean±SD change in VA was −4.0±15.1 ETDRS letters. The authors concluded that EMBT produces stable VA in most participants with previously treated disease, and may reduce the need for frequent anti-VEGF retreatments.129
The CABERNET study was a multicentric, randomised, active-controlled, phase 3 clinical trial with the same objective. There were 494 participants with treatment-naive neovascular AMD. Participants with classic, minimally classic, and occult lesions were randomised in a 2:1 ratio to an EMBT or a ranibizumab monotherapy control arm. Participants in the EMB arm received two mandated, monthly loading injections of 0.5 mg ranibizumab. The control arm received three mandated, monthly loading injections of ranibizumab and then quarterly injections. Both arms also received monthly as-needed (PRN) retreatment. At 24 months, 77% of the EMBT group and 90% of the control group lost fewer than 15 letters. This end point was non-inferior, using a 20% margin and a 95% CI for the group as a whole and for classic and minimally classic lesions, but not for occult lesions. Mean VA change was −2.5 letters in the EMBT arm and +4.4 letters in the control arm. At least one serious adverse event occurred in 54% of the participants in the EMB arm, most commonly postvitrectomy cataract, versus 18% in the control arm. Mild, non-proliferative radiation retinopathy occurred in 3% of the EMB participants, but in no case was it vision-threatening. The authors concluded that 2-year efficacy data did not support the routine use of EMB for treatment-naive patients with neovascular AMD, despite an acceptable safety profile.128
More recently, investigators have revisited external beam therapy, using a technique called stereotactic radiotherapy or radiosurgery. This treatment directs beams from different angles relative to the target area, thereby minimising the exposure to surrounding healthy tissue, and at the same time precisely targeting the radiation energy onto the lesion. The system is designed to overcome the traditional disadvantages of external beam therapy by dividing the dose into several separate beams that pass into the eye via different locations on the sclera.132 The patient is secured in position with a head restraint and the eye is continually tracked during treatment.131 In the INTREPID study, 230 patients with AMD, a history of a maximum of 3 years’ duration of CNV, and at least three anti-VEGF injections in the previous year were enrolled. Patients were randomised into three groups. The first group received 16 Gy irradiation, with a mandatory ranibizumab injection at the beginning of the study. The second group received 24 Gy irradiation and one ranibizumab injection as in the first group, while the control group received sham irradiation and one mandatory ranibizumab injection. All groups were allowed to have ranibizumab injections on an as-needed basis during the 12-month follow-up. At month 12, the primary end point of the study was met showing a 30% less need for anti-VEGF retreatment in the active compared with sham treatment group. Furthermore, a subgroup analysis of the patients revealed that patients with smaller lesions and high macular volume needed 55% fewer reinjections.133
Recommendation
At present, the only scientific argument to support the use of irradiation for the treating of neovascular AMD is the reduction in the number of retreatments necessary, but its delivery methods, efficacy and safety results are still controversial and need further investigations.
Surgical treatment for haemorrhagic AMD
Rationale
Subretinal haemorrhage (SRH) is a rare, but devastating complication of neovascular AMD.134 Damage to overlying photoreceptors has been found to occur within 24 h and degeneration of outer retinal layers within 3 days.117 The natural history of submacular haemorrhage (SMH) associated with neovascular AMD usually leads to poor VA. To date, intravitreal injections of anti-VEGF drugs have been the gold standard for the treatment of CNV secondary to AMD. Notwithstanding, clinical trials conducted for drug approval, such as the MARINA and ANCHOR studies of ranibizumab, excluded patients with predominantly haemorrhagic lesions. Several other studies that evaluated the benefits for haemorrhagic AMD of anti-VEGF drugs reported limited success in the patients affected.135 A complete mechanical elimination of the subretinal blood clot with or without extraction of the underlying neovacular complex is the rationale for submacular surgery in haemorrhagic AMD.
Evidence
Natural history studies found that 80% of patients had worsened vision, with a decline of mean VA from 20/240 to 20/1250: 38% developed fibrous tissue proliferation, 25% an atrophic scar and 22% an RPE rip.136 Decreased long-term visual outcome has been associated with an increased thickness of SMH, increased area of SRH and recurrent SMH.120 ,136 The use of antithrombotic medications was also associated with an increased risk of SMH, which can be of up to 11.6 times higher in patients with AMD.137
Shultz et al138 classified of SMH in a review article. A small haemorrhage was defined as a haemorrhage that does not extend to the vascular arcades, a medium haemorrhage as one that does extend to the vascular arcades, and a massive haemorrhage as one that extends past the vascular arcades to the periphery. A thin SMH was classified as less than 500 µ in thickness, and a thick one as more than 500 µ in thickness.
Many surgical modalities are available nowadays, including vitrectomy with manual clot extraction, vitrectomy with subretinal recombinant tissue plasminogen activator (rtPA), and pneumatic displacement with and without intravitreal rtPA. The treatment options for a small haemorrhage that does not extend to the vascular arcades are intravitreal anti-VEGF and pneumatic displacement with or without intravitreal rtPA.138
Several studies have investigated pneumatic displacement as an initial monotherapy to treat SMH and have reported successful displacement of haemorrhage with gas and prone positioning alone.139 ,140 Fibrin has been shown to form 1 h after experimentally created SRH in animal models. Therefore, rtPA has been used to liquefy SMH and to aid in its removal or displacement.141 Hassan et al reported on 14 patients with AMD and related SMH of less than 3 weeks duration, who were treated with intravitreal rtPA (25–100 µg), expansile gas and prone positioning. In all patients, the haemorrhage cleared within 5 days. By final follow-up, 67% of eyes improved by at least two Snellen lines.142 Hattenbach et al prospectively evaluated 43 eyes with AMD and related SMH with less than 1 month's duration. All patients were treated with intravitreal rtPA (50 µg) and sulfa hexafluoride followed by prone positioning. The best postoperative VA compared with preoperative VA improved by two or more Snellen lines in 19 eyes (44%) and remained stable in 24 eyes (56%). The authors noted that SMH of ≤14-days’ duration was associated with a better gain of lines of vision.
In 2007, Chen et al reported the results of a retrospective case series of 104 eyes that had received intravitreal injection of 100−30 µg of rtPA and expansile gas, and underwent prone positioning. In 64% of the eyes, the best VA improved at least two Snellen lines at the 3-month follow-up. The most common cause was AMD (86%), but the eyes with SMH unrelated to AMD had better VA outcomes.143
Medium-sized SMH extends to the vascular arcades and can be managed by either pneumatic displacement with or without intravitreal rtPA or PPV. In 1988, De Juan and Machemer were the first to perform PPV on four patients with AMD and SMH of 1-week to 1-year duration. All the operations were successful in removing SMH, but resulted in poor VA outcomes.144 Peyman and colleagues first described the use of subretinal rtPA (12.5 µg) as an adjuvant to PPV in three patients. They suggested that rtPA could reduce surgical manipulation of the retina and allow removal of the haemorrhage with smaller retinotomies. VA improved in one patient and was stabilised in the other two patients.145 Ibanez et al reported the results of a comparison between mechanical clot extraction with an extrusion cannula or forceps through a retinotomy and tPA-assisted drainage in 47 patients. No statistically significant differences in VA outcomes were found, with most patients having a final VA worse than 20/200.146 When rtPA was injected using a bent 36-gauge needle, and there was no waiting time for intraoperative clot lysis, all 11 eyes had clearance of SMH cleared in all 11 eyes, and 45% of eyes had a postoperative VA of 20/200 or better. VA improved compared with preoperative vision in 8 of 11 eyes with a mean follow-up of 6.5 months.147 SMH recurred in 27% of the eyes. The results of the Submacular Surgery Trials for predominantly haemorrhagic subfoveal CNV secondary to AMD were released in 2004. PPV was followed by removal of the entire lesion (including the CNV membrane, blood and scar tissue), subretinal rtPA at the surgeon's discretion (rtPA used in 38%; left for 40 min) and air or gas. The authors reported no benefit of submacular surgery relative to observation with respect to achieving stable or improved VA. However, they did report a reduced risk of severe vision loss (loss of ≥6 lines). Patients receiving surgery had higher rates of retinal detachment and cataract extraction compared with observation.148
Treating massive SMH is a challenge. The submacular surgery trials excluded patients with SMH greater than 9 disk areas.148 Oshima et al defined massive haemorrhage as extending to the periphery and involving at least two quadrants with haemorrhagic and bullous retinal detachment. In eight patients, intravitreal rtPA was injected 12–24 h prior to PPV, and eyes were examined for evidence of clot liquefaction followed by PPV. Heavy liquid was injected to displace haemorrhage through the retinotomies into the vitreous cavity for removal. The vitreous cavity was filled with perfluoropropane and the patient was instructed to remain face down for at least 24 h. Improvement in VA was seen in seven of eight patients and recurrence of SRH in one patient after 14 months.149
In a study with similar conditions, Fine et al examined 15 eyes that had undergone 20-gauge PPV and subretinal rtPA (25 µg) with a 39-gauge needle for 30–45 min. Four of the 15 patients had an inferior 180°–360° retinotomy during the primary procedure with excision of the clot with vitrectom or fragmatom. Patients received either long-acting gas (n=12) or silicone oil (n=3), and were positioned semiprone after the operation for 2 weeks. Nine of the 15 patients required other surgical interventions. Although the improvement in VA at 1 year was modest, the authors stated that it was favourable compared with the natural history.150 Complications associated with vitrectomy include retinal detachment, proliferative vitreoretinopathy, epiretinal membrane, RPE tear, progression of cataract, vitreous haemorrhage, creation of a macular hole, and recurrence of SMH.148
As the use of anti-VEGF drugs for neovascular AMD is well established, their use combined with PPV and subretinal rtPA has also been investigated. Treumer et al injected bevacizumab into the subretinal space of 12 patients followed by PPV and subretinal rtPA (10–20 µg). Air-fluid exchange was performed and patients were positioned prone for 1 day. Displacement of SMH was achieved in 75% of patients and mean improvement of VA was logMAR 0.96 at 12 weeks.151 Arias et al reported results of PPV with intravitreal bevacizumab in 15 eyes. Subretinal rTPA was injected for thick SMH and intravitreal rTPA for thin SMH (at the end of surgery). They reported that all patients had improved VA at final follow-up.152
Recommendation
As there is no consensus on the optimal treatment, and data from studies often conflict, no general recommendation can be given for the treatment of haemorrhagic AMD. A classification of SMH based on size appears practical. Pneumatic replacement may be useful in small haemorrhages, while medium-sized clots may require a surgical approach including vitrectomy. Massive haemorrhage has a genuinely unfavourable prognosis. Attention should be given to the fellow eye to prevent active disease, and a haemorrhagic event in the eye that sees better. Evidence levels are generally low with levels II–III.
Acknowledgments
The authors thank Andreas Pollreisz, MD for critical review and preparation of the manuscript.
References
Footnotes
-
Contributors Concept and design of the article (US-E, GR); drafting of manuscript (US-E, VC, AL, ML, ES, RS, BE, JM, FB); critical review and revision of article (US-E, VC, AL, ML, ES, RS, BE, JM, GR, FB).
-
Competing interests Alcon Laboratories. (US-E, VC, AL, ML, ES, RS, BE, JM, GR, FB); Bayer Healthcare (US-E, VC, AL, ML, ES, RS, BE, JM, GR, FB); Novartis (US-E, VC, AL, ML, ES, RS, BE, JM, GR, FB); Boehringer (US-E); Allergan (FB).
-
Ethics approval All studies referred to in this review article have properly undergone ethics approval procedures.
-
Provenance and peer review Not commissioned; externally peer reviewed.