Article Text

Abstract
Background/Aim To evaluate efficacy and safety of bimatoprost 0.03% preservative-free (PF) ophthalmic solution versus bimatoprost 0.03% (Lumigan) ophthalmic solution for glaucoma or ocular hypertension.
Methods In this double-masked, parallel-group study, patients were randomised to bimatoprost PF or bimatoprost for 12 weeks. The primary analysis for non-inferiority was change from baseline in worse eye intraocular pressure (IOP) in the per-protocol population at week 12. For equivalence, it was average eye IOP in the intent-to-treat population at each time point at weeks 2, 6 and 12.
Results 597 patients were randomised (bimatoprost PF, n=302 and bimatoprost, n=295). The 95% CI upper limit for worse eye IOP change from baseline was <1.5 mm Hg at each week 12 time point, meeting prespecified non-inferiority criteria. The 95% CI upper limit for the treatment difference for average IOP was 0.69 mm Hg and the lower limit was −0.50 mm Hg at all follow-up time points (hours 0, 2 and 8 at weeks 2, 6 and 12), meeting equivalence criteria. Both treatments showed decreases in mean average eye IOP at all follow-up time points (p<0.001), were safe and well tolerated.
Conclusions Bimatoprost PF is non-inferior and equivalent to bimatoprost in its ability to reduce IOP-lowering with a safety profile similar to bimatoprost.
- Glaucoma
- Treatment Medical
- Ocular surface
- Intraocular pressure
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Bimatoprost, a prostamide introduced in 2001,1 ,2 has a profound intraocular pressure (IOP)-lowering effect in patients with ocular hypertension (OHT) and open-angle glaucoma.3 Administered once daily in these patients, bimatoprost 0.03% ophthalmic solution (Lumigan; Allergan, Irvine, California, USA) has shown greater efficacy in lowering IOP in clinical trials and meta-analyses4–6 than prostaglandin analogues such as latanoprost7 ,8 and travoprost.8 ,9 Similar to prostaglandin analogues, common side effects include conjunctival hyperaemia, increased iris pigmentation, eyelash growth and periocular skin pigmentation, with most being mild.10
As with other multiuse ophthalmic medications, bimatoprost 0.03% ophthalmic solution contains a preservative to maintain sterility, specifically benzalkonium chloride at 50 parts per million. While benzalkonium chloride, the most commonly used preservative in topical β-blockers and prostaglandin agonists, has a safety record spanning decades of use, a small percentage of patients may be sensitive to the preservative.11 This study evaluated the safety and efficacy of a preservative-free (PF) bimatoprost 0.03% formulation that was developed as an alternative for patients with sensitivity or allergies to preservatives.12
Methods
Study design and participants
This was a prospective, multicentre (36 sites in the USA), double-masked, randomised, parallel-group, 12-week study (ClinicalTrials.gov Identifier: NCT01099774) to compare the efficacy and safety of new PF bimatoprost 0.03% ophthalmic solution (bimatoprost PF) and bimatoprost 0.03% ophthalmic solution (bimatoprost) administered once daily for 12 weeks in patients with OHT or glaucoma. The study was conducted in compliance with applicable Good Clinical Practice guidelines. Study methods did not change after enrolment began.
Eligible patients were at least 18 years old with OHT, chronic open-angle glaucoma, chronic-angle closure glaucoma with patent iridotomy/iridectomy, pseudoexfoliative glaucoma or pigmentary glaucoma in both eyes. At baseline, after 4-day to 4-week washout of IOP-lowering medications, patients were required to have an IOP of 22–30 mm Hg in each eye with asymmetry between eyes of no more than 3 mm Hg, and best-corrected visual acuity equivalent to a Snellen score of 20/100 or better in each eye. The minimum washout period was 4 days for parasympathomimetics and topical or systemic carbonic anhydrase inhibitors, 2 weeks for sympathomimetics and α-agonists, and 4 weeks for β-adrenergic blocking agents, combination products and prostaglandin agonists. Exclusion criteria included uncontrolled systemic disease (ie, any systemic disorder such as respiratory, cardiac, hepatic, endocrine or renal disease that is unstable or uncompensated); allergy or sensitivity to any component of the study medication; central corneal thickness >600 μm or <500 μm; recent or anticipated alteration of existing chronic systemic medications that could affect IOP (eg, systemic β blockers); anterior segment laser or other intraocular surgery within 6 months; chronic use of ocular medications other than study medications (occasional use of artificial tears was allowed but not within 24 h prior to a visit); use of contact lenses during the study; intermittent use of systemic corticosteroids within 21 days before any study visit; use of ophthalmic corticosteroids within 2 months or during the study; history of inadequate IOP control on bimatoprost monotherapy; and ocular surface findings at baseline such as trace or greater hyperaemia or irritation. Patients who were pregnant, nursing or who could become pregnant during the study were excluded.
Treatment and assessments
Eligible patients were stratified by baseline mean diurnal IOP (≤24 mm Hg or >24 mm Hg) and randomly assigned by an automated interactive voice/web response system to receive bimatoprost PF or bimatoprost in a 1:1 ratio. Allergan Biostatistics prepared the randomisation schedule and each investigator was assigned blocks for each IOP stratum. The formulations differed only in the absence of benzalkonium chloride in bimatoprost PF. Patients were dispensed study medication kits containing unit-dose containers (each for single use) that were identical for both formulations. Patients were instructed to instil one drop in each eye once daily in the evening, starting between 19:00 and 21:00 on the day of the baseline visit. Follow-up visits were scheduled at weeks 2, 6 and 12.
IOP was measured using a Goldmann applanation tonometer and a two-person reading method13 at 8:00, 10:00 and 16:00 at each visit. IOP was measured twice in each eye; if results differed by >1 mm Hg, a third measurement was taken. IOP for each eye was determined as the mean of two or median of three readings. Diurnal IOP was defined as the mean of all time points at a visit.
Safety was evaluated by adverse events (AEs, coded using the Medical Dictionary for Regulatory Activities V.14.0), biomicroscopy, fundus examination (including cup/disc ratio), macroscopic hyperaemia graded by gross inspection in comparison with standard photographs, visual acuity, visual field measurement, photographic iris colour assessments and vital signs.
Endpoints and analyses
Two sets of analyses were performed, based on worse eye IOP and average eye IOP, to address regulatory requirements of different countries. The worse eye refers to the eye with the higher mean diurnal IOP at the baseline visit. If both eyes had the same mean diurnal IOP at baseline, the right eye was designated as the worse eye. In the worse eye analysis, the primary efficacy analysis was change from baseline at each hour evaluated at week 12 using the per-protocol (PP) population (patients without a major protocol violation) based on a priori guidance sought from the European Regulatory authorities. Treatments were compared using an analysis of covariance (ANCOVA) model with treatment and investigator as main effects and baseline worse eye IOP as covariate. Bimatoprost PF would be considered non-inferior to bimatoprost if the upper limit of the 95% CI of the between-group difference did not exceed 1.5 mm Hg at any hour at week 12.
Secondary efficacy analyses included change from baseline in worse eye IOP at week 12 using the intent-to-treat (ITT) population (all randomised patients). The treatments would be considered equivalent if the 95% CI upper limit was ≤1.5 mm Hg and the lower limit was ≥1.5 mm Hg at each hour. Statistical significance (α=0.05) of within-group change from baseline was used in the final analysis for each time point separately. The proportion of responders, defined as patients with at least a 20% reduction in worse eye IOP from the corresponding baseline hour at week 12, was analysed using Pearson's χ2 or Fisher's exact test. The worse eye mean diurnal IOP was analysed with CIs based on an ANCOVA model with treatment and investigator as main effects and baseline worse eye mean diurnal IOP as covariate. Estimated difference (bimatoprost PF minus bimatoprost) was based on least squares means from the ANCOVA model.
In the average eye analysis (across both eyes), the primary efficacy analysis was average IOP at each hour at weeks 2, 6 and 12 in the ITT population. Missing data were imputed using the last observation carried forward. A two-sided 95% CI for the treatment difference (bimatoprost PF minus bimatoprost) was constructed from an analysis of variance model with fixed effects of treatment and investigator. Bimatoprost PF would be equivalent to bimatoprost if the upper and lower limits of the 95% CI of the between-group difference were within the 1.5 mm Hg margin at each follow-up time point and within 1.0 mm Hg at the majority of time points. The estimated treatment difference based on least squares means was also determined. Because the requirement was to meet set margins at specified time points, no adjustment to significance level was required for multiple time points.
A secondary efficacy analysis was change from baseline in average IOP at each follow-up time point in the ITT population. The methods, analysis of variance model and criteria for declaring equivalence were the same as for the primary analysis.
Subgroups were analysed by baseline characteristics of age (≤65 years or >65 years), sex (male or female), race (black, non-black (Caucasian, Asian, Hispanic or other)), and iris colour (light irides (blue, blue-grey, grey, green, hazel or other light) and dark irides (blue/grey-brown, green-brown, brown, dark brown or other dark)) for worse eye and average eye analyses.
Sensitivity analyses were conducted for worse eye and average eye by actual value and change from baseline in the ITT population with missing values imputed using multiple imputation methods, adjusting for baseline and without adjusting for investigator. Imputed data were analysed using an ANCOVA model with treatment as fixed effect and baseline average or worse eye IOP as covariate. A two-sided 95% CI for the treatment difference was constructed from the ANCOVA model.
Sample size calculations considered a one-sided α=0.025, 90% power with no expected between-group differences. Based on a maximum SD of 3.74 mm Hg at week 12, 132 patients were required to demonstrate non-inferiority at a given hour. For the equivalence test of average eye IOP in the ITT population, based on a maximum SD of 3.03 mm Hg with the equivalence limit of ±1.5 mm Hg or ±1.0 mm Hg, 108 or 240 patients per treatment, respectively, were required to demonstrate equivalence at a given time point. The largest sample size from the three estimates was required for adequate power for all criteria and therefore, 240 patients per treatment group were required. The study was to randomise 534 patients based on an expected 10% dropout rate.
Results
Between June 2010 and April 2011, 597 patients were randomised (see online supplementary figure S1); 98.0% completed the study. Patient demographics and baseline characteristics were similar between the treatment groups (table 1).
Demographics and baseline characteristics, intent-to-treat population
Worse eye analysis
Bimatoprost PF was non-inferior to bimatoprost for change from baseline in worse eye IOP at each hour at weeks 2, 6 and 12 in the PP population. The 95% CI upper limit of the between-group difference did not exceed 0.78 mm Hg at any hour (table 2). Both treatments showed statistically and clinically significant decreases in IOP from baseline at all time points (p<0.001) (figure 1). Mean change from baseline ranged from −7.49 mm Hg to −5.93 mm Hg for bimatoprost PF and −7.77 mm Hg to −6.06 mm Hg for bimatoprost (table 2). The between-group difference in mean worse eye IOP was <0.4 mm Hg at all time points and was not statistically or clinically significant (see online supplementary figure S2).
Mean±SD change from baseline in worse eye intraocular pressure (mm Hg), per-protocol population
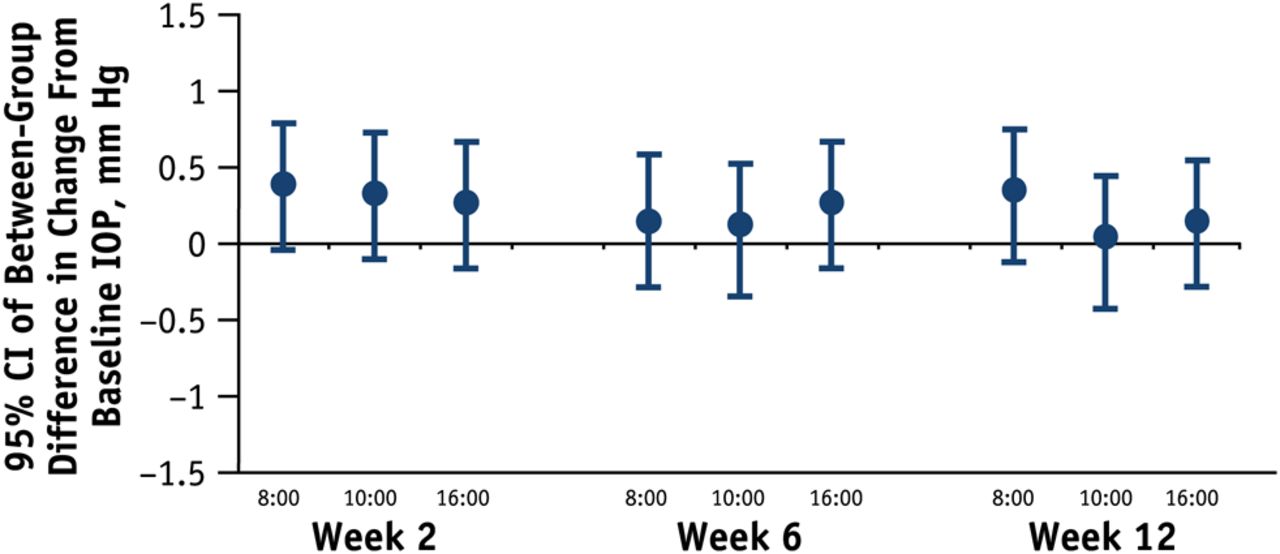
Between-group differences at each time point in change from baseline in worse eye intraocular pressure (IOP), per-protocol population. Upper limit of 95% CI of between-group difference ≤0.75 mm Hg at week 12.
In the ITT population, the upper and lower 95% CI limits for mean change from baseline in worse eye IOP at week 12 were within a 1.0 mm Hg margin at each hour, and within the prespecified 1.5 mm Hg margin defined as equivalence. Both treatments showed statistically and clinically significant mean decreases at all time points (p<0.001).
The proportion of ITT responders at week 12 ranged from 70.2% to 80.8% for bimatoprost PF and 69.5% to 82.0% for bimatoprost (p>0.05 for between-group differences) (see online supplementary table S1).
Among the other analyses, the mean between-group difference in worse eye mean diurnal IOP ranged from 0.13 mm Hg to 0.28 mm Hg across follow-up visits (see online supplementary table S2). There were no clinically meaningful differences in the subgroup analyses. Sensitivity analyses showed results similar to the primary analyses.
Average eye analysis
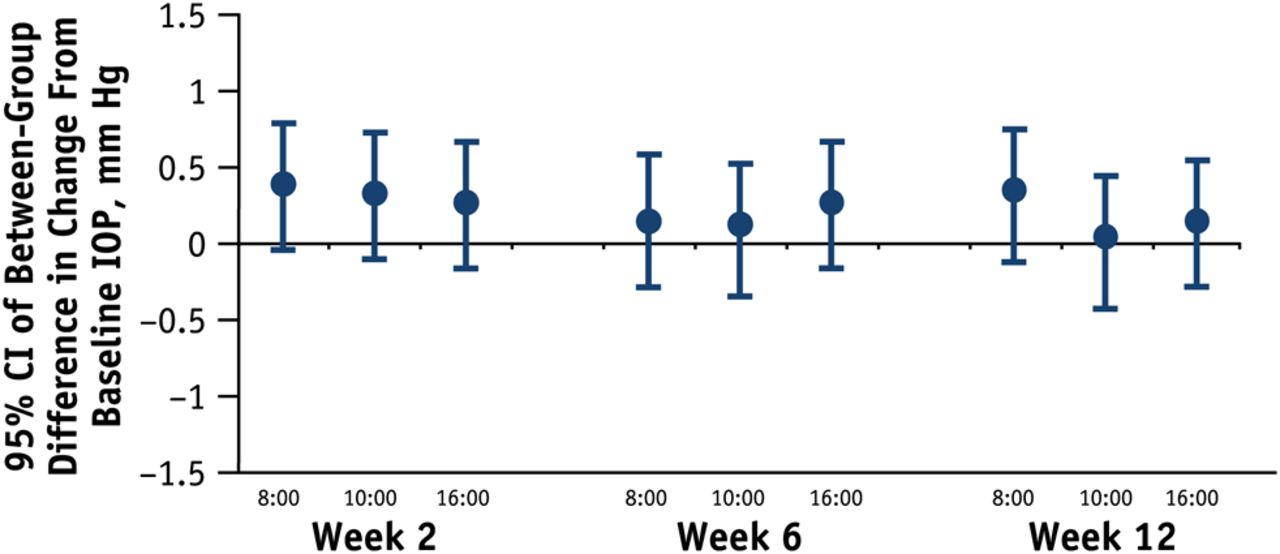
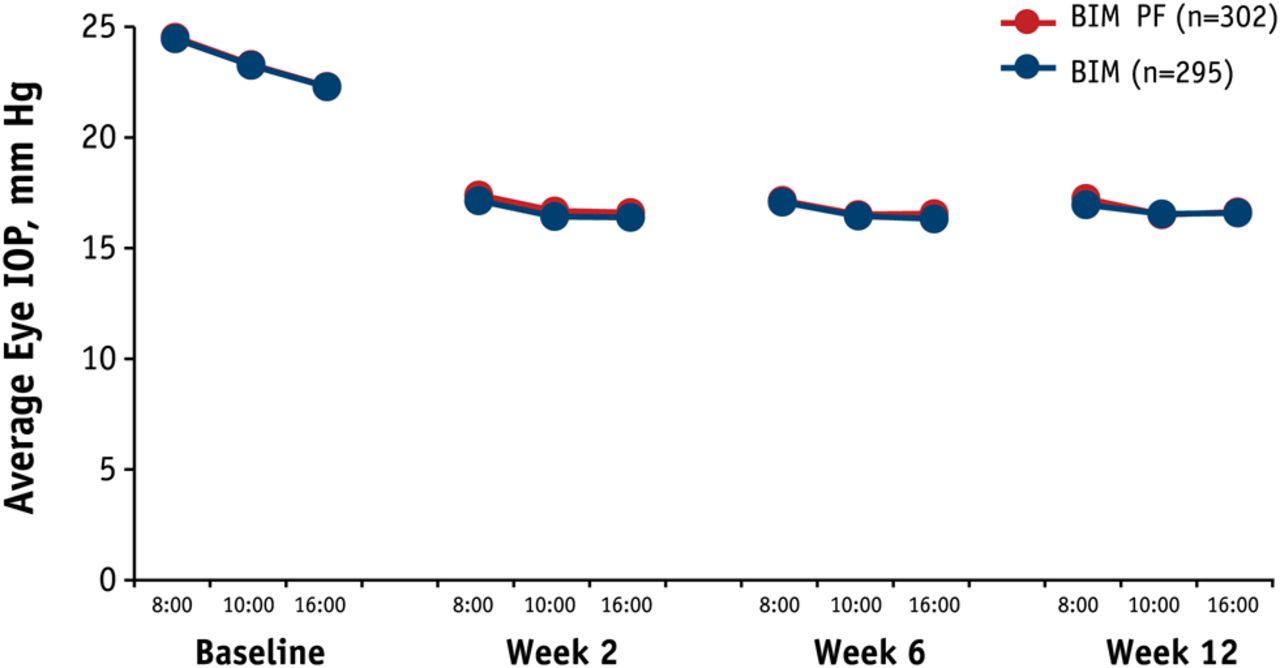
Bimatoprost PF was equivalent to bimatoprost in average eye IOP at all follow-up time points in the ITT population. The 95% CI upper limit of the between-group difference did not exceed 0.69 mm Hg and the lower limit was not lower than −0.50 mm Hg. There were no statistically or clinically significant between-group differences in the ITT population (figure 2). Mean treatment differences (bimatoprost PF minus bimatoprost) ranged from −0.07 mm Hg to 0.25 mm Hg. Results were similar for the PP population.

{kind=link}
{kind=link}
Mean average intraocular pressure (IOP) at each time point, intent-to-treat population. Difference between groups <0.3 mm Hg. Note the lines overlay making them somewhat indistinguishable. BIM, bimatoprost 0.03% ophthalmic solution (blue); BIM PF, bimatoprost 0.03% preservative-free ophthalmic solution (red).
For mean change from baseline in average eye IOP at all time points in the ITT population, the upper and lower 95% CI limits were within a 1.0 mm Hg margin at each hour, which was the prespecified margin defined as equivalence. Both treatments showed statistically and clinically significant mean decreases from baseline at all time points (p<0.001). Mean change from baseline ranged from −7.36 mm Hg to −5.67 mm Hg and −7.50 mm Hg to −5.70 mm Hg for bimatoprost PF and bimatoprost groups, respectively. Subgroup and sensitivity analyses results were similar to those for the worse eye analyses.
Safety and tolerability
Both treatments were well tolerated. AEs were reported for 40.5% (122/301) of bimatoprost PF and 44.1% (130/295) of bimatoprost patients (p=0.382), with ocular AEs reported for 31.9% and 34.9%, respectively (p=0.434). The most frequent ocular AE was conjunctival hyperaemia (table 3), which was considered to be treatment-related in all except one patient in each group. Conjunctival hyperaemia AEs were reported as mild or moderate for the majority of patients. For patients who had undergone washout of prostaglandin agonists, treatment-related conjunctival hyperaemia was reported for 18% (35/194) of bimatoprost PF and 20.7% (35/169) of bimatoprost patients, compared with 33.6% (36/107) and 32.5% (41/126), respectively, of patients who had not undergone washout from prostaglandin agonists.
Patients with ocular adverse events ≥2% in either group, safety population
AEs other than ocular were reported for 13.6% (41/301) of bimatoprost PF and 14.2% (42/295) of bimatoprost patients (p=0.828), with nasopharyngitis being the most common. Skin hyperpigmentation AEs, all of which were considered to be treatment-related, were reported for 1.0% (3/301) of bimatoprost PF and 0.7% (2/295) of bimatoprost patients.
Six patients (bimatoprost PF, n=2 and bimatoprost, n=4) reported serious AEs, including one patient with metastatic urothelial cancer who died; no serious AE was ocular or considered to be treatment-related (see online supplementary table S3). Five patients (bimatoprost PF, n=2 and bimatoprost, n=3) discontinued due to AEs. Treatment-related AEs that led to discontinuation (some patients had more than one) were conjunctival hyperaemia (three patients), foreign body sensation in eyes (two patients), eye pruritus, drug hypersensitivity and eye irritation (one patient each).
The most common finding on biomicroscopy was increased severity (grade ≥1) of conjunctival hyperaemia, which was reported for 35.0% (105/301) of bimatoprost PF and 36.6% (108/295) of bimatoprost patients (p=0.660); most cases were trace or mild (see online supplementary figure S3). The maximum grade on macroscopic hyperaemia evaluations was similar for both treatments; the majority were trace or mild. Increased severity (grade ≥1) for punctate keratitis was more frequent with bimatoprost than bimatoprost PF (6.8% (20/295) and 3.7% (11/301), respectively; p=0.086); most cases were trace or mild with one severe case in the bimatoprost PF group at week 12.
Iris colour changed (based on photographs) for 0.3% (1/301) of bimatoprost PF and 1.0% (3/295) of bimatoprost patients (p=0.369). There were no statistically or clinically significant between-group differences for change from baseline in cup/disc ratio, best-corrected visual acuity at week 12 or visual field. There were no clinically significant between-group differences in vital signs.
Discussion
PF ophthalmic formulations are needed for patients with OHT or glaucoma who have known sensitivity to preservatives.12 ,14 For those patients, the bimatoprost PF formulation provides a well-tolerated and efficacious treatment alternative.
Treatment in patients with OHT or glaucoma is aimed at lowering IOP to preserve visual function. The impact of IOP reduction on disease progression has been well established.15–19 While many IOP-lowering medications are available the choice of therapy must consider IOP-lowering ability, tolerability and other factors.20 The PF formulation of tafluprost 0.0015% (Saflutan; Merck Sharp & Dohme Corp., Whitehouse Station, New Jersey, USA) is currently the only approved PF prostaglandin, and has been shown to be non-inferior to timolol in IOP lowering.21 An earlier study comparing a preserved formulation of tafluprost with latanoprost 0.005% did not meet the prespecified non-inferiority requirement.22 In a recent study, patients with primary open-angle glaucoma previously treated with latanoprost, travoprost or bimatoprost for at least 3 months and having ocular discomfort were switched to PF tafluprost. There were no differences in mean daily IOP with tafluprost compared with latanoprost and travoprost, but mean daily IOP was statistically significantly lower with bimatoprost than tafluprost (p<0.05). The severity scores for conjunctival hyperaemia and punctate keratitis were significantly higher with bimatoprost 0.03% than tafluprost but mean scores for both treatments were in the mild range.23
This trial comparing bimatoprost PF versus bimatoprost was analogous to the comparison of PF tafluprost and PF timolol in its randomised, double-blind, multicentre non-inferiority design.22 More recently, a PF formulation of latanoprost has been developed. A 3-month single-masked trial24 compared the efficacy and safety of this new latanoprost formulation with the benzalkonium chloride-preserved latanoprost. Patients entering the trial had been successfully treated with latanoprost (IOP ≤18 mm Hg) for at least 9 months. The trial concluded the non-inferiority of PF latanoprost to the preserved formulation, the authors suggesting a better local tolerance with the former. The selection of patient population should be taken into account when assessing tolerability and when comparing across studies that use different populations.
The 12-week time frame of this study was its major limitation. Studies of longer duration and those that evaluate tolerability in patients receiving multiple topical medications and in subgroups of patients such as those with severe ocular surface disease are warranted.
Clinical trials and meta-analyses4–6 have shown bimatoprost 0.03% ophthalmic solution to provide efficacious treatment for OHT and glaucoma, demonstrating greater reductions in IOP compared with prostaglandin analogues such as latanoprost7 ,8 and travoprost.8 ,9 In the present study, bimatoprost 0.03% PF demonstrated non-inferiority and equivalence in IOP lowering when compared with bimatoprost 0.03%, with no significant between-group differences in safety and tolerability. Bimatoprost 0.03% PF thus provides an efficacious IOP-lowering alternative for patients with sensitivity to preservatives.
Acknowledgments
Medical writing assistance was provided by Linda Whetter, DVM, PhD, of Evidence Scientific Solutions and Jean Siegel, PhD, and was funded by Allergan.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online tables
Footnotes
-
Contributors DGD and MB are guarantors for this paper.
-
Funding The study and medical writing support were sponsored by Allergan, Irvine, California, USA. The study sponsor participated in the study design, analysis and interpretation of the data, in writing the report and in the decision to submit the paper for publication.
-
Competing interests DGD has received support from Allergan for research, travel, advisory board membership, manuscript development/review and is a consultant and lecturer; TRW has no conflicts to declare; GFS is a consultant and lecturer for Allergan; TKM is a lecturer for Allergan; MB, CL and RMS are employees of Allergan.
-
Ethics approval Schulman Associates Institutional Review Board, (Cincinnati, Ohio, USA), Biomedical Research Associates of New York, Investigational Review Board (Lake Success, New York, USA) and Western Institutional Review Board (Olympia, Washington, USA).
-
Provenance and peer review Not commissioned; externally peer reviewed.