Article Text

Abstract
Background/aim To assess long-term effects of dexamethasone intravitreal implant (DEX implant) monotherapy on retinal morphology in diabetic macular oedema (DME).
Methods Two multicentre, masked, phase III studies with identical protocols randomised patients with DME, best-corrected visual acuity of 34–68 Early Treatment Diabetic Retinopathy Study letters and central subfield retinal thickness (CSRT) ≥300 µm to DEX implant 0.7, 0.35 mg or sham procedure. Patients were followed up for 3 years (39 months if treated at month 36), with retreatment allowed at ≥6-month intervals. Patients needing other macular oedema (ME) therapy exited the study. Changes from baseline in CSRT, macular volume and ME grade, area of retinal thickening, macular leakage, macular capillary loss and diabetic retinopathy severity were assessed.
Results After 3 years, more eyes treated with DEX implant 0.7 and 0.35 mg than sham showed improvement (although small) in ME grade (p<0.05 vs sham). DEX implant 0.7 mg delayed time to onset of two-step progression in diabetic retinopathy severity by ∼12 months. DEX implant 0.7 and 0.35 mg produced small, non-sustained reductions in macular leakage but had no significant effect on macular capillary loss.
Conclusions DEX implant 0.7 or 0.35 mg, administered at ≥6-month intervals over 3 years, produced sustained retinal structural improvement in DME.
Trial registration number NCT00168337 and NCT00168389.
- Anatomy
- Clinical Trial
- Drugs
- Imaging
- Retina
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Over the last decade, new therapeutic approaches have resulted from improved understanding of the pathophysiological processes responsible for endothelial blood–retinal barrier breakdown in diabetic macular oedema (DME).1 Several inter-related ocular inflammatory events are of particular relevance: the release of vascular permeability factors such as vascular endothelial growth factor (VEGF); upregulation of inflammatory mediators; increased expression of endothelial adhesion molecules and the influx and adhesion of leucocytes to the retinal microvasculature (leukostasis), resulting in endothelial cell injury and apoptosis.2–5 Several VEGF inhibitors, including aflibercept, bevacizumab, pegaptanib and ranibizumab, have shown clinical efficacy as intravitreal therapies for DME.6 ,7 Current treatment guidelines recognise the role of ranibizumab in improving visual acuity in patients with macular centre involvement and vision loss due to DME.8 However, per-protocol clinical use of VEGF inhibitors in DME requires monthly intravitreal injections.8 Moreover, many patients exhibit retinal thickening despite anti-VEGF therapy, highlighting the need for additional treatments.
Intravitreal corticosteroids downregulate expression of cytokines such as tumour necrosis factor-α,9 nuclear factor-κB,9 VEGF9 and intercellular adhesion molecule-1,9 ,10 and inhibit leukostasis and retinal microvasculature leakage.9 ,10 Fluocinolone acetonide (Retisert, Bausch & Lomb, Bridgewater, New Jersey, USA; Iluvien, Alimera Sciences, Alpharetta, Georgia, USA) and dexamethasone (DEX implant; Ozurdex, Allergan, Irvine, California, USA) are available commercially as slow-release intravitreal implant systems. DEX implant has recently been approved for the treatment of adults with DME. In early-phase clinical trials in DME, DEX implant monotherapy11 and combination laser therapy12 were effective in improving central subfield retinal thickness (CSRT) and best-corrected visual acuity (BCVA). These findings have recently been confirmed in two pivotal phase III trials of DEX implant in DME (the MEAD studies). In a pooled analysis of the MEAD study data, DEX implant met the primary efficacy end point of ≥15-letter improvement in BCVA and showed acceptable tolerability.13 The anatomical findings from the MEAD studies are detailed here.
Methods
Study design and participants
Data collected from two identically designed, 3-year, multicentre, masked, phase III trials (NCT00168337 and NCT00168389) of the safety and efficacy of DEX implant in the treatment of DME (MEAD trials) were pooled. The trials were conducted at 131 sites in 22 countries worldwide between February 2005 and June 2012. The protocol was approved by the institutional review board/ethics committee at each study site, and the trials were conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent. The protocol is described in detail elsewhere13 and is summarised below.
Adults with diabetic retinopathy–associated macular oedema involving the fovea, previously treated with medical or laser therapy, and an Early Treatment Diabetic Retinopathy Study (ETDRS) BCVA in the study eye of 34–68 letters (20/200–20/50) were enrolled. Treatment-naïve patients refusing laser treatment or who, in the investigator's opinion, would not benefit from laser treatment also were eligible. Retinal thickness in the central 1 mm macular subfield measured by time-domain optical coherence tomography (TD-OCT; Stratus OCT3 or OCT2 (used for only 1% of study submissions), Carl Zeiss Meditec, Dublin, California, USA) was required to be ≥300 µm in the study eye. Key exclusion criteria were uncontrolled diabetes (glycosylated haemoglobin >10%); glaucoma; ocular hypertension (untreated intraocular pressure >23 mm Hg); aphakia; active iris or retinal neovascularisation; history of pars plana vitrectomy or steroid-induced ocular hypertension; recent intraocular laser or incisional surgery or intravitreal VEGF inhibitor or triamcinolone treatment; and current use of systemic steroids.
Patients were randomised (1:1:1) to intravitreal DEX implant 0.7 mg, DEX implant 0.35 mg or a sham procedure in the study eye. If both eyes were eligible, the eye with the shorter duration of macular oedema was selected. Patients were evaluated for retreatment every 3 months during the 3-year study, but retreatment could not be performed more often than every 6 months. Retreatment was allowed if retinal thickness in the 1 mm central macular subfield was >225 µm (revised to >175 µm or evidence of residual oedema in a protocol amendment in 2010). Patients needing adjunctive or other therapy for macular oedema were required to exit the study prior to administration of additional treatment. Efficacy data captured before patient exit were included in the analysis.
Study assessments and end points
The primary efficacy end point in the MEAD trials was the percentage of patients with ≥15-letter improvement in BCVA from baseline in the study eye at final assessment (end of year 3 or 39 months for patients treated at month 36). Prespecified secondary end points included changes in retinal anatomy measured using OCT, fundus photography and fluorescein angiography. Image evaluation (grading) was performed at a centralised reading facility (University of Wisconsin Fundus Photograph Reading Center, Madison, Wisconsin, USA) by certified masked technicians.
TD-OCT (Stratus OCT3 or, if unavailable, OCT2) was conducted at 3-month intervals. Six radial scans, each ∼6 mm long and centred on the fovea and performed using fast macular thickness map settings (128 A-scans/B-scan), were supplemented by high-resolution 6 mm cross-hair scans (512 A-scans/B-scan). Stereoscopic 30° or 35° colour fundus photographs of the study eye were taken at baseline, every 3 months during the first year and every 6 months during the second and third years. Fundus photographs were assessed for presence and extent of retinal thickening, diabetic retinopathy severity level and presence of clinically significant macular oedema (CSME). Diabetic retinopathy was graded using the ETDRS Final Retinopathy Severity Scale condensed to nine severity categories.14 Outcomes of interest included changes from baseline in disc area of central retinal thickening and macular oedema grade (improvement, no change or worsening).
Fluorescein angiography was performed at baseline, months 6, 12 and 24 and at the end of year 3 (or 39 months for patients treated at month 36) to assess macular fluorescein leakage and perifoveal capillary integrity. Grading protocols were adapted from the ETDRS clinical trials.15 The mean change from baseline to study end in total disc area of macular capillary loss and the proportions of patients with and without ischaemia (defined as a total area of macular capillary loss >0.5 disc area) at baseline and the last visit were determined. Details of the assessments are described in the online supplementary materials.
Statistical analyses
Unless stated otherwise, all efficacy analyses were performed with missing values imputed by last observation carried forward (LOCF) for the intent-to-treat (ITT) population (all randomised patients). Treatment comparisons and estimates based on LOCF were supported by sensitivity analyses using multiple imputation. Area under the curve (AUC) analysis of the average change from baseline in CSRT during the study used observed values in the ITT population; missing values were not imputed. Areas of central retinal thickening, fluorescein leakage and macular capillary loss and changes from baseline in CSRT and macular volume were analysed using analysis of covariance with baseline value as a covariate. Changes from baseline in proportions of patients with CSME and central retinal thickening were compared using Wilcoxon rank-sum test. Proportions of patients in each diabetic retinopathy severity category and proportions of patients with ≥2-step progression from baseline in diabetic retinopathy severity category were analysed using the Cochran–Mantel–Haenszel general association test stratified by study. Time to two-step progression in diabetic retinopathy severity category was analysed using the Kaplan–Meier method, and cumulative progression rates were compared using the log-rank test. All statistical tests were two-sided and performed at α=0.05 significance level. SAS V.9.3 (SAS Institute, Cary, North Carolina, USA) was used.
Results
Study population
The pooled ITT population comprised 1048 randomised patients (table 1), of whom 607 patients (57.9%) completed all visits. Completion rates were appreciably higher in the DEX implant 0.7 mg (64.1%) and 0.35 mg (66.3%) groups than in the sham group (43.4%).
Baseline demographic and study eye characteristics of the ITT population
The median number of study treatments administered in each DEX implant group ranged between 4 and 5 compared with 3 in the sham group. Baseline demographics and study eye characteristics did not differ significantly among the three treatment groups (table 1).
OCT findings
DEX implant–treated eyes showed marked fluctuation in the reduction in CSRT at consecutive study visits, particularly during year 1, creating a saw-tooth pattern of treatment effect (figure 1; <1% of scans were deemed non-gradable). Similar results were obtained with the observed data analysis. Study eyes treated with DEX implant showed greater reductions from baseline in CSRT than sham-treated eyes at all time points (figure 1).

Mean change from baseline in retinal thickness in the central subfield versus time. p≤0.024 at all time points for dexamethasone intravitreal (DEX) implants versus sham (analysis of covariance with treatment and study as fixed effects and baseline value as covariate). B, baseline; F, final visit.
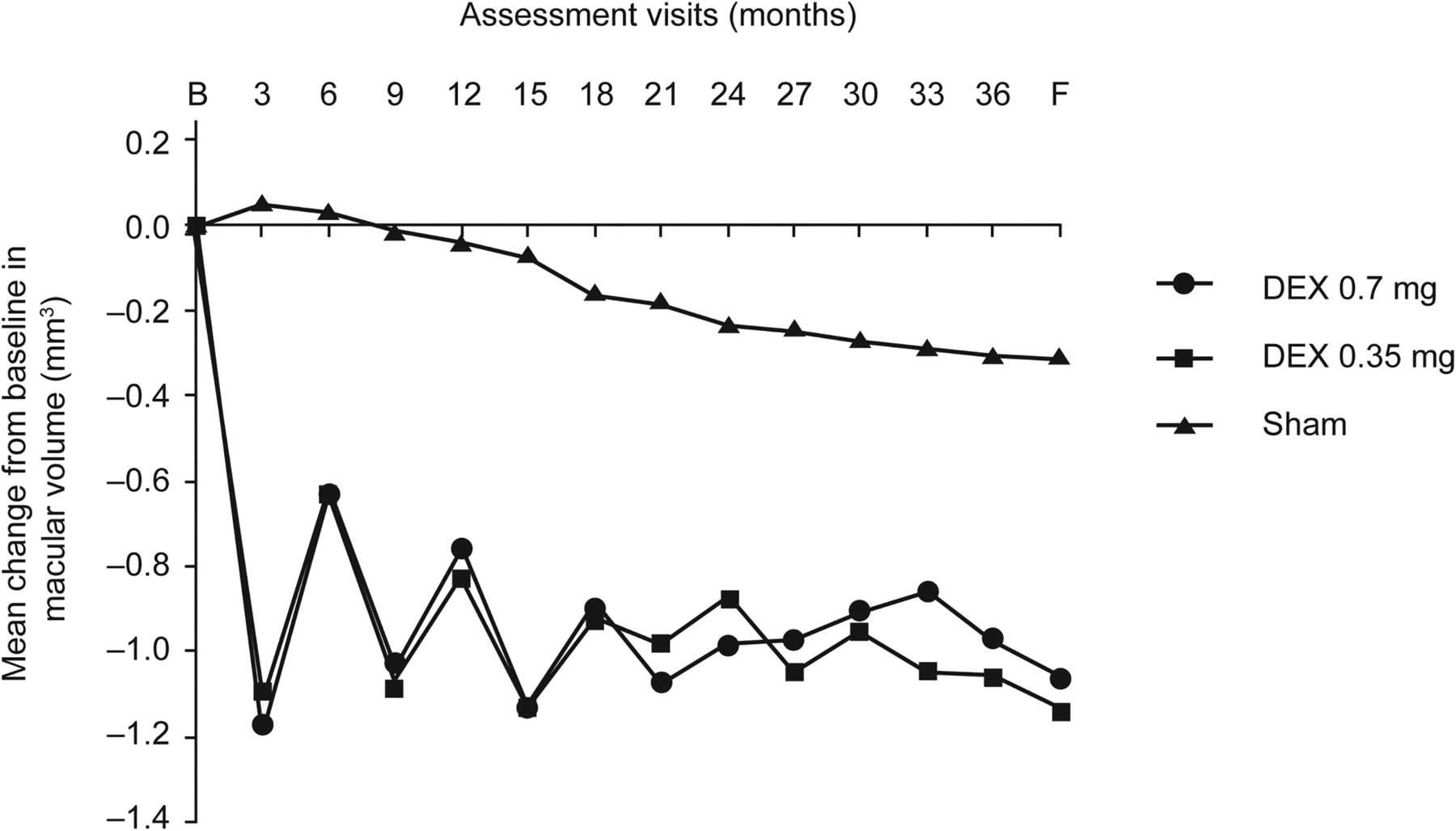
At the final study visit, CSRT was reduced by (mean) 117.3 and 127.8 μm in DEX implant 0.7– and 0.35 mg–treated eyes versus 62.1 μm in sham-treated eyes (both p<0.001 vs sham; table 2). Over the full study period, the mean average reduction in CSRT (AUC approach) was greater in DEX implant–treated eyes (table 2). At all study time points from month 3 onward, the proportion of study eyes with CSRT >250 μm was lower in the DEX implant than the sham treatment groups. At the final study visit, the decline in the proportion of study eyes in this category was greater with DEX implant 0.7 mg (from 94.5% (baseline) to 60.2%) and DEX implant 0.35 mg (from 94.8% to 58.7%) than with sham (from 95.9% to 71.6%). Likewise, at all study time points DEX implant–treated eyes displayed greater reductions from baseline in macular volume than sham-treated eyes (both doses p<0.001 vs sham at study end; figure 2 and table 2).
Summary of optical coherence tomography, fundus photography and fluorescein angiography findings

{kind=link}
{kind=link}
Mean change from baseline in macular volume versus time. p≤0.002 at all time points for dexamethasone intravitreal (DEX) implants versus sham (analysis of covariance with treatment as a fixed effect and baseline value as covariate). B, baseline; F, final visit.
Fundus photography findings
The area of central retinal thickening showed greater reduction from baseline in DEX implant-treated versus sham-treated eyes at all study time points; at study end, mean reductions of 2.75 and 2.93 disc areas were recorded in DEX implant 0.7– and 0.35 mg–treated eyes, respectively (both p<0.001 vs sham; table 2). The corresponding absolute area of central retinal thickening at study end was (mean) 5.34, 5.38 and 6.13 disc areas, respectively.
Compared with sham-treated patients, DEX implant-treated patients showed a delay of ∼12 months in onset of two-step progression in diabetic retinopathy severity (10th percentile of time to progression was ∼36 months for both the DEX implant 0.7 mg and 0.35 mg treatment groups vs ∼24 months for the sham treatment group; p=0.03 and p=0.08, respectively). The 10th percentile of time to two-step improvement in diabetic retinopathy severity was ∼24 and ∼13 months for the DEX implant 0.7 and 0.35 mg treatment groups versus ∼24 months for sham (p=0.655 and p=0.364, respectively).
The prevalence of CSME declined steadily in both DEX implant treatment groups, and more gradually in the sham group. At study end, fewer DEX implant 0.7– and 0.35 mg–treated eyes had CSME than sham-treated eyes (p<0.05 and p<0.01, respectively; table 2). More DEX implant 0.7– and 0.35 mg–treated eyes showed an improvement in CSME (shift from a higher to a lower grade) between baseline and study end than sham-treated eyes (p<0.05 and p<0.01, respectively; table 2).
Fluorescein angiography findings
DEX implant 0.7 mg– and DEX implant 0.35 mg–treated eyes showed greater reductions (from baseline) in total area of macular fluorescein leakage than sham-treated eyes during the first year (mean −0.35 and −0.52 vs +0.11 disc areas at 6 months, both p<0.05 vs sham; mean −0.58 and −0.79 vs −0.03 disc areas at 12 months, p<0.01 for DEX implant 0.35 mg vs sham). However, these effects were modest and were not sustained; at study end the reductions in total area of fluorescein leakage in the DEX implant treatment groups were not significantly different from those of the sham group (table 2). Changes (from baseline) in total area of macular capillary loss did not differ significantly between DEX implant– and sham-treated eyes at any time (table 2).
Discussion
This pooled analysis of data from the MEAD trials13 is the first to examine the long-term retinal changes associated with the visual acuity improvements produced by intravitreal DEX implant in DME. DEX implant, administered with a median frequency of four to five injections over a 3-year period, provided sustained improvements in OCT and fundus photography–based anatomical markers of macular oedema, whereas macular fluorescein leakage showed improvement only over the first year. Compared with sham-treated eyes, DEX implant–treated eyes displayed significant decreases in CSRT, macular volume and central retinal thickening at all study time points, with treatment differences emerging as early as month 3. There was no significant effect on macular capillary loss.
The present findings extend the limited anatomical information provided by earlier short-term phase II studies of DEX implant 0.7 and 0.35 mg in patients with DME.12 ,16 ,17 In keeping with previous studies suggesting that the efficacy of DEX implant on CSRT peaks at approximately 1–3 months before gradually declining,11 ,12 ,16 the profile of mean change in CSRT versus time observed in the present study was characterised by a saw-tooth pattern (figure 1), with each cycle of improvement presumably corresponding to retreatment with DEX implant. A similar saw-tooth pattern was obtained with observed data. This pattern is most likely explained by the gradual decline over time in DEX release from the implant in situ, and suggests that a retreatment interval of less than 6 months may be required for a more consistent anatomical response.
The reduction in macular fluorescein leakage noted during year 1 is consistent with findings from previous short-term, phase II studies.12 ,16 ,17 However, the reason for the discrepancy between the longer-term OCT and fluorescein angiography findings is unclear. Although fluorescein angiography is a reliable method for qualitative assessment of fluid leakage, a reduction in fluorescein intensity is not always accompanied by a reduction in area of fluorescein leakage. Likewise, fluorescein angiography findings do not always mirror OCT findings in DME: in some cases, macular leakage may be evident in the absence of an increase in retinal thickness; in other cases, fluorescein angiography may fail to detect intraretinal or subretinal fluid that is evident on OCT.18 Additionally, the diffuse fluorescein staining of non-cystoid oedema in DME may be below the detection threshold of the OCT instrument.18 ,19
Strengths of the present study include its large patient population, its extended duration and inclusion of a sham treatment arm, investigator and patient masking to treatment and a range of end points. The study design ensured that treatment outcomes were not complicated by the effects of adjunctive DME therapies. However, the requirement for subjects requiring adjunctive treatment to exit the study resulted in high attrition, particularly in the control group, which may have adversely affected ITT/LOCF analyses. Other potential study limitations include the lack of adjustment for glycaemic and blood pressure control, which may affect macular thickness,20 and the lower reproducibility of macular thickness measurements obtained with TD-OCT, the standard technology when the study was initiated (2004), compared with current spectral-domain OCT instruments.21 Variability is also likely to have arisen from substitution of centre-point thickness for CSRT owing to scan quality issues and possibly from the natural diurnal variation in macular thickness in eyes with DME.22 Nevertheless, manual centre-point thickness measurements performed by the reading centre operators showed excellent reproducibility.
In summary, this pooled data analysis extends the evidence from previous studies in DME, indicating that DEX implant 0.7 and 0.35 mg provides sustained improvements in anatomical measures of macular oedema over a 3-year treatment period. DEX implant thus has the potential to reduce the need for laser therapy and provides an alternative to anti-VEGF therapy in DME.
Acknowledgments
Writing and editorial assistance was provided to the authors by Andrew Fitton, PhD, of Evidence Scientific Solutions (Horsham, UK). The full list of participating investigators is published in the primary manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors RPD and X-YL are guarantors for this paper. All authors participated in the study design, data interpretation and drafting, critical review, revision and approval of the manuscript. HC conducted the data analysis. The study sponsor participated in the design of the study, data analysis and data interpretation and preparation, review and approval of the manuscript.
Funding The study and medical writing support were sponsored by Allergan, Inc., Irvine, California, USA.
Competing interests RPD and SS have received grant support and consulting fees from Allergan, Inc. X-YL and YH are employees of Allergan, Inc. At the time of the study, HC and SMW were employees of Allergan, Inc.
Ethics approval As this was a multicentre study, several IECs and IRBs approved the study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement For additional unpublished data from the study, Allergan should be contacted.
Linked Articles
- At a glance