Article Text

Abstract
Aims To demonstrate non-inferiority of ranibizumab treat-and-extend (T&E) with/without laser to ranibizumab pro re nata (PRN) for best-corrected visual acuity (BCVA) in patients with diabetic macular oedema (DMO).
Methods A 24-month single-masked study with patients randomised 1:1:1 to T&E+laser (n=121), T&E (n=128) or PRN (control; n=123). All patients received monthly injections until BCVA stabilisation. The investigator decided on re-treatment in the PRN and treatment-interval adaptations in the T&E groups based on loss of BCVA stability due to DMO activity. Likewise, laser treatment was at investigator's discretion. Collectively, these features reflect a real-life scenario. Endpoints included mean average change in BCVA from baseline to months 1–12 (primary), mean BCVA change from baseline to months 12 and 24, treatment exposure and safety profile.
Results Both T&E regimens were non-inferior to PRN based on mean average BCVA change from baseline to months 1–12 (T&E+laser: +5.9 and T&E: +6.1 vs PRN: +6.2 letters; both p<0.0001). Mean BCVA change at month 24 was similar across groups (+8.3, +6.5 and +8.1 letters, respectively). The mean number of injections was 12.4 and 12.8 in the T&E+laser and T&E groups and 10.7 in the PRN group. The T&E regimens showed 46% reduction in the number of clinic visits. Over 70% of patients maintained their BCVA, with treatment intervals of ≥2 months over 24 months. Safety profile was consistent with that described in the product information.
Conclusions T&E is a feasible treatment option for patients with DMO, with a potential to reduce treatment burden. Slightly more injections were required versus PRN, likely due to the specifics of the T&E regimen applied here.
Trial registration number NCT01171976.
- Vision
- Clinical Trial
- Macula
- Treatment Medical
- Treatment Lasers
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Diabetic macular oedema (DMO) is the most common cause of permanent vision loss in working-age adults with diabetes.1–3 Patients with DMO represent a heterogeneous group with varied responses to therapy that have led to individualised dosing regimens of antivascular endothelial growth factors. Currently, clinicians often practise a pro re nata (PRN) approach, wherein patients are observed monthly and treated upon signs of disease activity, or a treat-and-extend (T&E) approach, which allows incremental increase in treatment intervals with an aim to identify the longest possible treatment and visit-free interval for a given patient. The effectiveness of a PRN regimen in DMO has been established with ranibizumab 0.5 mg (Lucentis®; Genentech, South San Francisco, California, USA; and Novartis Pharma AG, Basel, Switzerland) in the long-term RESTORE and DRCR.net (protocol I) studies. In these studies, the initial best-corrected visual acuity (BCVA) improvements observed at year 1 were maintained through years 2, 3 and 5, with a reduced number of injections.4–9 However, a PRN regimen tends to require frequent clinic visits to monitor disease status and administer treatment if needed.
The T&E approach was first introduced by Spaide and Freund in 2007 for neovascular age-related macular degeneration (nAMD), with an aim to reduce patients’ treatment burden by individualising treatment intervals and reducing the number of clinic visits.10 Studies have shown that individualised T&E regimens improve visual outcomes in nAMD and require fewer injections than those administered in a monthly regimen and fewer monitoring visits than those in a PRN regimen.11–15
Although the DRCR.net (protocol I) study demonstrated that DMO can be managed with less than monthly monitoring and longer treatment intervals7–9 and the recent RELIGHT study demonstrated that bimonthly monitoring intervals were feasible in maintaining initial visual acuity (VA) gains over 12 months,16 no T&E regimen has been evaluated in patients with DMO prior to RETAIN, the first prospective study designed to evaluate a T&E regimen in the management of DMO. The merits of two T&E regimens (with/without laser therapy) were assessed by comparing directly with the established PRN regimen. The ranibizumab PRN regimen was as per the European Summary of Product Characteristics (EU SmPC, 2011).17 Here, we report the 24-month outcomes from the RETAIN study.
Materials and methods
Between September 2010 and April 2013, 372 patients with visual impairment due to DMO were enrolled at 64 centres across 13 European countries (list of investigators available in online supplementary S1) in this 24-month, phase IIIb, single-masked (VA assessor and patient were both masked to treatment assignment), controlled, three-arm parallel-group study. Written informed consent was obtained from each participating patient before study entry. RETAIN (registered at http://www.ClinicalTrials.gov; NCT01171976) adhered to the tenets of the Declaration of Helsinki, the International Conference on Harmonisation and Good Clinical Practice guidelines.
Patient eligibility and study treatment
The inclusion and exclusion criteria of RETAIN were comparably broader than previous confirmatory studies in DMO and aimed at inclusion of a population with relevance for real life. Patients aged >18 years with either type I or II diabetes mellitus (defined per American Diabetes Association or WHO guidelines) with glycosylated haemoglobin (HbA1c) values of ≤12% at screening and an Early Treatment Diabetic Retinopathy Study (ETDRS) BCVA letter score ranging from 78 to 39, inclusive (approximate Snellen equivalent of 20/32–20/160), those with visual impairment due to focal or diffuse DMO18 of any extent or thickness in at least one eye who were eligible for laser treatment in the opinion of the investigator, were eligible for inclusion. One eye was treated as the study eye. If both eyes were eligible, the eye with worse VA was selected as the study eye.
Patients were excluded if they showed structural damage within 0.5 disc diameter of the centre of the macula in the study eye likely to preclude improvement in VA following the resolution of macular oedema; BCVA >73 letters and central subfield thickness (CSFT) <300 μm in the study eye; any intraocular surgery in the study eye within 3 months prior to randomisation; history of vitrectomy in study eye regardless of time prior to randomisation; panretinal and focal/grid laser photocoagulation in the study eye within 6 and 3 months prior to randomisation; treatment with antiangiogenic drugs in either eye (pegaptanib sodium, anecortave acetate, bevacizumab, ranibizumab, vascular endothelial growth factor (VEGF)-Trap) within 3 months prior to randomisation; active intraocular inflammation in either eye (grade trace or above); any active infection in either eye (conjunctivitis, keratitis, scleritis, uveitis or endophthalmitis); and uncontrolled glaucoma in either eye (intraocular pressure >24 mm Hg on medications or per investigator's judgement). The complete list of exclusion criteria is presented in online supplementary S2. No additional exclusions were applied by the investigators during screening.
The patients were randomised (1:1:1) to receive either ranibizumab 0.5 mg T&E with laser (T&E+laser; n=121), ranibizumab 0.5 mg T&E without laser (T&E; n=128) or ranibizumab 0.5 mg PRN (PRN (control); n=123; online supplementary figures S1A and S1B). Details regarding patient randomisation are available in online supplementary file S3. All three treatment groups received monthly ranibizumab 0.5 mg until BCVA was stabilised (no change in BCVA over three consecutive months with treatment). In the ranibizumab T&E+laser group, patients received laser treatment on day 1, after which laser could be readministered based on the ETDRS guidelines. Laser treatment was at the discretion of the investigator, reflecting a real-life scenario, with a 3-month minimum interval recommended between treatments (see online supplementary figure S1B and file S4).
T&E design
The T&E regimen allowed the incremental extension of intertreatment intervals based on disease stability; VA loss due to disease recurrence triggered a return to monthly injections until VA stability was re-established. This conservative approach was chosen owing to lack of experience with T&E in DMO when the RETAIN study was designed. For the same reason, the maximal length of an intertreatment interval was capped at 3 months.
In detail, patients in all treatment groups received monthly ranibizumab 0.5 mg injections for at least three consecutive months until BCVA stability was achieved. BCVA stability judgement was at the discretion of the assessing clinician with no prespecified criteria. At the visit when BCVA stability was recorded, no treatment was administered. Patients randomised to either of the T&E groups were then scheduled for treatment at the next visit, that is, the treatment interval was extended to 2 months. If the patient's vision remained stable after these two months, the treatment interval was extended to 3 months, and this interval length was maintained till the patient's vision remained stable (figure 1). During treatment intervals of >1 month, patients continued study visits during the intervening months solely to maintain masking, that is, no treatment was given and no adaptation of the intertreatment interval was allowed. For PRN group patients, monthly monitoring visits were scheduled after initial confirmation of BCVA stability, and treatment reinitiated by loss of VA due to disease activity. Further details on study visits, laser treatment and re-treatment criteria are provided in online supplementary file S4.

The treat-and-extend (T&E) treatment algorithm. *Scheduled between the T&E visits where no study treatment was administered and no decision for study treatment was made. For the pro re nata (PRN; control) regimen, each monitoring visit was also a potential treatment visit. †Best-corrected visual acuity (BCVA) stable: no BCVA improvement or deterioration noted for three consecutive monthly study visits under treatment. **Patient's BCVA worsened due to diabetic macular oedema (DMO) disease activity. ¶First visit followed 1 month after the visit at which stabilisation (at month 3) was confirmed. BSL, baseline; M, months.
Objectives
The primary objective was to demonstrate non-inferiority (four-letter margin) of the T&E regimen with/without laser to the PRN regimen with respect to mean average change in BCVA from baseline to month 1 through month 12. If non-inferiority was established, superiority of the T&E regimens was evaluated. Secondary objectives included the evaluation of the mean average change in BCVA from baseline to month 1 through month 24; mean change in BCVA and change in CSFT (average retinal thickness of the circular area with 1 mm diameter around the foveal centre) from baseline to months 12 and 24; mean number and pattern of treatments over 12 and 24 months; impact of laser on the number of re-treatments in the T&E groups and incidence of ocular and non-ocular adverse events (AEs) and serious AEs (SAEs).
Study assessments and analysis
Best-corrected visual acuity
A certified evaluating investigator (masked to the treatment assignment) used ETDRS-like VA testing charts at a starting distance of 4 m to assess BCVA of the study eye. Additionally, this investigator performed other study efficacy assessments and judged the presence or absence of BCVA stability and disease activity or recurrence.
Optical coherence tomography
Spectral or time-domain optical coherence tomography (OCT) was performed at every study visit. The same device was to be used for a given patient throughout the study. All images were assessed by trained and qualified experts at the sites; no central reading centre was involved. Change in CSFT was analysed as change from baseline in per cent.
Treatment exposure
The number of ranibizumab injections and laser treatments were recorded for each treatment group. Other endpoints included average treatment interval, that is, the interval (in months) between visits at which treatment was administered to the study eye, from the first treatment visit after initial BCVA stability was confirmed up to month 24, and the total number of visits scheduled for treatment after the visit with initial BCVA stability up to month 24.
Safety
Safety assessments included the incidence of ocular and non-ocular AEs and SAEs, their frequency and relationship to treatment/ocular injection. All AEs were summarised by system organ class, based on the preferred term, and were grouped per the standardised Medical Dictionary for Regulatory Activities.
Statistical analysis
A sample size of 104 patients per treatment group had >90% power to establish non-inferiority at a four-letter margin for at least one of the two T&E regimens compared with the PRN regimen in terms of the mean average change in BCVA based on a one-sided significance level of 0.0125, assuming a treatment difference of 1 letter, SD of 10 letters and underlying normal distribution for an unstratified Mann–Whitney test. The primary analysis was conducted after patients had completed the month 12 visit using the full analysis set (FAS), which comprised all randomised patients who received at least one application of the study treatment (ranibizumab or laser) and had at least one postbaseline BCVA assessment. The mean value/last observation carried forward approach was used to impute missing postbaseline data. Patients were analysed according to the treatment assigned at randomisation (intent-to-treat principle). Hypotheses of the primary objective for non-inferiority and superiority were tested using a sequentially rejective multiple testing procedure that protects the multiple one-sided alpha level of 0.025.19 The safety analysis was conducted on the safety set that comprised all patients who received at least one application of study treatment and had at least one postbaseline safety assessment. Patients were assigned to treatment groups according to the actual treatment they received. Further details are provided in online supplementary file S5.
Results
Patient demographics and baseline characteristics
Of the 372 enrolled patients, 332 (89.2%; mean age, 63.7 years; males, 62.4%) completed the study. The efficacy analysis was performed on the FAS (n=359), and the safety analysis on the safety set (n=370; online supplementary figure S2).
AEs and withdrawal of consent were the most common reasons for discontinuation across all treatment groups (see online supplementary figure S2). Overall, patient demographics as well as baseline disease and ocular characteristics were generally well balanced across treatment groups (table 1). Patients had mild to moderate vision loss at baseline (mean BCVA: 63.4 letters; WHO, International Classification of Functioning, Disability and Health; online supplementary file S6). Further details are provided in table 1.
Key patient demographics and baseline diabetes and ocular characteristics (randomised set)
Efficacy
Best-corrected visual acuity
The primary endpoint was met, with both the T&E regimens (T&E ranibizumab+laser and T&E ranibizumab) being non-inferior to PRN with respect to mean average change (±SD) in BCVA from baseline to month 1 through month 12 (5.91 (±5.53) letters and 6.14 (±5.71) letters vs 6.20 (±6.01) letters, respectively; online supplementary figure S3 and table S2). The upper limits of the 97.5% CIs for differences in least-squares mean for the PRN versus T&E groups were <4 letters for both comparisons. The superiority of T&E groups over the PRN group was not established (see online supplementary figure S3 and table S2).
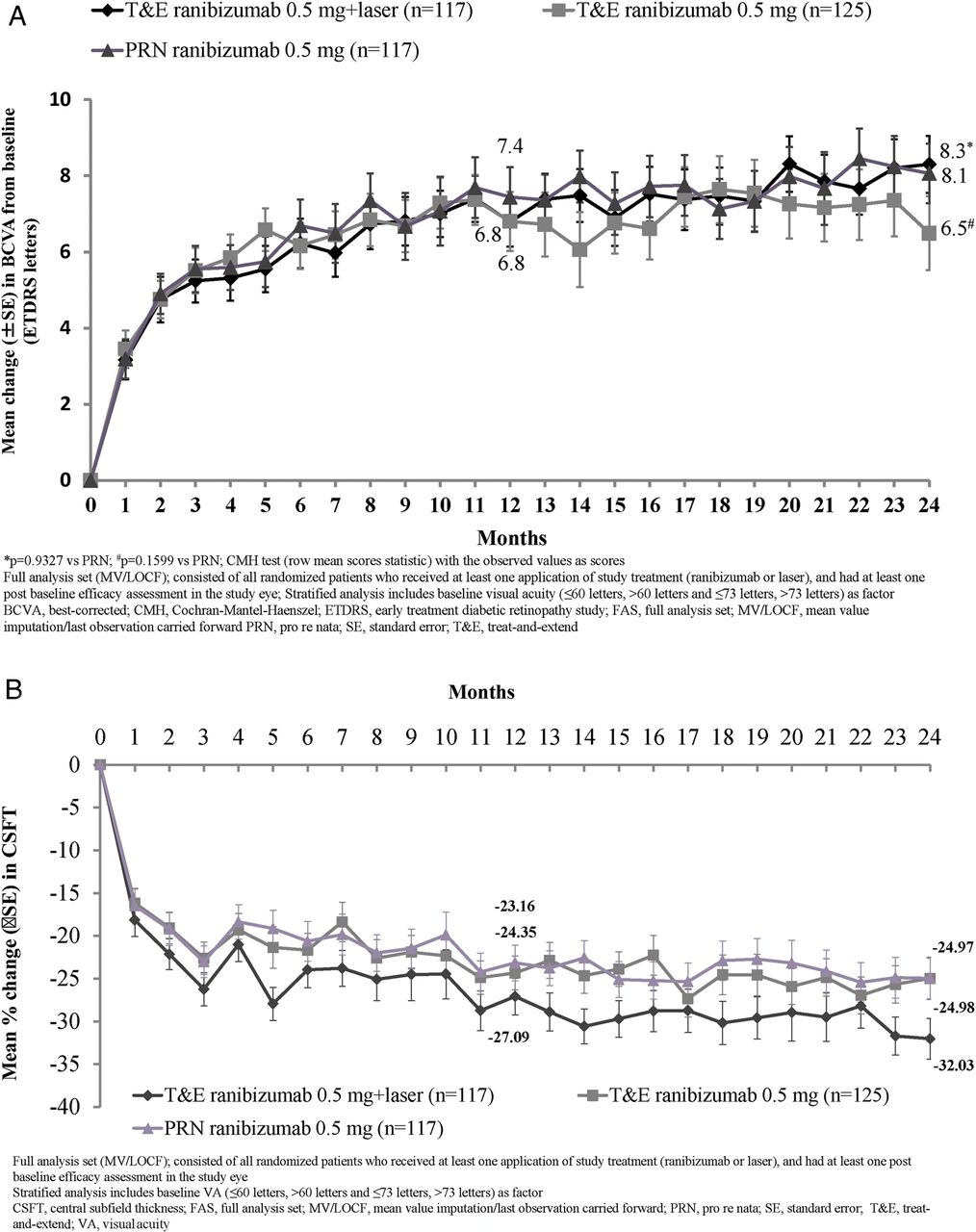
There was no statistical difference between the two T&E groups in terms of the average change in BCVA from baseline to month 1 through month 12 and 24 treatment periods (see online supplementary figure S3 and table S2). In all treatment groups, the mean BCVA increased from baseline during the first four months of treatment by ∼five letters, with subsequent steady increase of 1–3 letters over the following 20 months. At month 24, mean BCVA change from baseline improved across all treatment groups (figure 2A and table 2).
BCVA and CSFT outcomes at months 12 and 24 (FAS-MV/LOCF)
{kind=link}
{kind=link}
(A) Mean change in best-corrected visual acuity (BCVA) from baseline to months 12 and 24 (full analysis set (FAS)-mean value imputation/last observation carried forward (MV/LOCF)). *p=0.9327 versus pro re nata (PRN); #p=0.1599 versus PRN; Cochran–Mantel–Haenszel (CMH) test (row mean scores statistic) with the observed values as scores. (B) Mean percentage change in central subfield thickness (CSFT) from baseline over time (FAS-MV/LOCF). In (A) and (B), FAS (MV/LOCF) comprised all randomised patients who received at least one application of study treatment (ranibizumab or laser) and had at least one postbaseline efficacy assessment in the study eye. Stratified analysis included baseline visual acuity (≤60 letters, >60 and ≤73 letters and >73 letters) as factors. ETDRS, Early Treatment Diabetic Retinopathy Study.
Treatment exposure
Ranibizumab 0.5 mg injections and laser treatment
Over the initial 12 months, all treatment groups received a median of seven injections; over 24 months, median number of injections was 12 in both T&E groups and 10 in the PRN group. A majority of T&E+laser group patients (77.8%) received only one laser treatment over 24 months (table 3).
Number of ranibizumab injections and laser treatments, number of visits scheduled for treatment from months 3 to 24 and treatment intervals up to month 24 (safety set)
Average interval between treatments
After the visit with initial BCVA stability up to month 24, >70% patients in the T&E groups maintained their initial BCVA stability with intertreatment intervals of ≥2 months (table 3).
Number of visits scheduled for treatment
After the visit with initial BCVA stability up to month 24, the mean number of scheduled treatment visits was 9.0 and 8.9 for the T&E groups (with/without laser, respectively) and 16.6 for the PRN group (table 3).
Safety profile
Adverse events
Non-ocular and ocular AEs were reported in approximately 70% and 39% patients, respectively, across all treatment groups. The most frequent non-ocular and ocular AEs are listed in online supplementary table S1, with a majority being of mild to moderate intensity. Discontinuations from the study due to ocular and non-ocular AEs are shown in online supplementary table S2.
Serious adverse events
The overall incidence of ocular and non-ocular SAEs was low and similar across all treatment groups. Ocular SAEs in the study eye were reported in three patients in the T&E groups (vitreous haemorrhage and endophthalmitis in two T&E+laser group patients and periorbital haematoma in one T&E group patient), and no ocular SAEs were reported in the PRN group (see online supplementary table S3). Overall, at least one non-ocular SAE was reported in 85 patients during the study (33, 29 and 23 in the T&E+laser, T&E and PRN groups, respectively). The most commonly reported non-ocular SAEs are listed in online supplementary table S3. Most of the non-ocular SAEs were not suspected to be related to study drug and/or ocular injection.
Overall, seven deaths were reported: two (1.6%) in the T&E+laser group, four (3.2%) in the T&E group and one (0.8%) in the PRN group. Only two deaths were suspected to be treatment-related by the investigator (myocardial infarction in the T&E+laser group and cerebrovascular accident in the PRN group); details are provided in online supplementary table S3.
Discussion
Findings from the RETAIN study show that both T&E regimens, with/without laser, were non-inferior to a PRN regimen and resulted in improvement and maintenance of VA in patients with DMO over 24 months. Overall, efficacy findings in patients with mild to moderate vision loss at baseline (mean VA: 63.4±11.15 letters) in RETAIN were consistent with findings from the RESTORE study, which included a similar patient population in terms of baseline VA (63–65 letters in both ranibizumab groups with/without laser).4 A retrospective analysis from 1616 patients with DMO across nine phase II or III randomised clinical trials (eg, RETAIN, RESTORE, RIDE, RISE, VIVID, VISTA) of ranibizumab 0.5 mg and aflibercept 2 mg has demonstrated that, regardless of the dosing regimen and anti-VEGF compound, the mean VA at 12 months plateaued at about 70 (68.5–73.0) letters. This analysis revealed that greater BCVA gains are observed in patients with poor vision at baseline and vice versa. While in this comparative analysis the RESTORE and RETAIN trials demonstrated the lowest gain (approximately seven letters) in BCVA for each of the PRN and T&E arms, the mean baseline BCVA in RESTORE and RETAIN trials featured among the highest. In contrast, patients in RIDE and RISE had the lowest baseline BCVA but achieved the highest BCVA gains of about 12.0 letters with monthly ranibizumab 0.5 mg. Though these BCVA gains in RIDE and RISE were reported at 24 months, they were comparable to gains observed at 12 months (Dugel PU, Hillenkamp J, Sivaprasad S, et al. Unpublished work. Baseline visual acuity strongly predicts visual acuity gain in patients with DME following anti-VEGF treatment across trials in DME. Retina). Thus, considering baseline BCVA effects and the apparent ceiling effect observed for anti-VEGF therapy of DMO, the BCVA results across all three regimens applied in RETAIN are comparable to results obtained with monthly regimen or other anti-VEGF agents.
The T&E in RETAIN was associated with a slightly higher number of injections (mean 12.4 and 12.8 over 24 months for the T&E+laser and T&E groups, respectively) compared with the PRN group (10.7). This increase was most likely due to the constraints of the protocol: (1) over 24 months, the T&E regimen required a minimum of 10 injections versus a minimum of three injections with PRN; (2) the maximal treatment-free interval was capped at 3 months in the T&E regimen, but the treatment intervals could have been extended to beyond 3 months for certain patients, as indicated by 18% patients who had at least one interval of >3 months in the PRN group; and (3) any unsuccessful attempt to extend the treatment-free interval required the patient to return to monthly injections, which is more conservative than the T&E regimen described previously for nAMD.11–15 ,20 This conservative approach was selected when the RETAIN protocol was designed so as not to put patients with DMO at risk of vision loss. Today, a stepwise reduction of the treatment interval for patients with disease recurrence would be preferred over returning to monthly treatment, and the maximal interval would likely be extended to >3 months. Nevertheless, the T&E regimens in this study led to a substantial reduction of ∼46% in the number of clinic visits up to month 24 versus the PRN regimen.
Importantly, in a clinical setting, the treatment visit with T&E regimen is predefined; thus, the patient is aware of receiving the treatment at the next clinic visit, can prepare mentally for treatment and make adjustments in their schedule if necessary. Moreover, the clinic can pre-prepare the theatre schedule, injections and staffing, thus optimising time and resources.
In the RETAIN study, the frequency distribution of the average interval between treatment visits showed that a T&E regimen adapts to a patient's needs. During the 24-month study period, the proportion of patients who had an average interval between treatments of 2 and 3 months from initial BCVA stabilisation ranged from 36% to 44% in the T&E+laser group and 39–44% in the T&E group (table 3). Thus, in the absence of DMO activity, ∼44% patients in the T&E groups were able to maintain vision and extend the treatment-free interval up to 3 months. In fact, over 70% patients in the T&E groups maintained their BCVA improvement with intertreatment intervals of ≥2 months after the visit with initial BCVA stability up to month 24. Similarly, data from the DRCR.net (protocol I),8 ,9 READ-221 and the recent RELIGHT16 studies indicate that extended monitoring intervals are possible without a negative impact on BCVA outcomes.
In RETAIN, combining laser with T&E ranibizumab did not provide additional improvements in BCVA outcomes and did not affect the number of injections needed, consistent with findings from the DRCR.net (protocol I)8 ,9 and RESTORE studies.4 ,6 The majority of patients in the T&E+laser group (77.8%) received a single laser treatment at baseline and did not require any additional through 24 months; the number of laser treatment in RETAIN was similar to that observed in protocol I.8 ,9 The effects of laser may vary based on differing practices of laser administration among investigators. Findings in the T&E ranibizumab and PRN groups for efficacy and safety outcomes were consistent with those observed with the T&E ranibizumab with laser group in the study.
Patients with diabetes are at a higher risk of comorbidities and systemic complications.22 ,23 However, no new safety risks were identified with ranibizumab 0.5 mg used in the RETAIN study. Overall, the incidence of ocular/non-ocular AEs was low and consistent with that observed in the previously reported clinical studies of ranibizumab in DMO.4 ,6 ,8
To our knowledge, RETAIN is the first prospective study designed to evaluate a T&E regimen in the management of DMO. The study was single-masked considering that the treating investigator would see the laser burns and patients with prior laser experience can distinguish true laser from sham laser treatments. The study protocol was conservative with respect to T&E to ensure patients do not lose vision. Overall, the inclusion and exclusion criteria of RETAIN were inclusive and re-treatment decisions and decisions on laser treatment were largely based on the investigator's judgement, reflecting a real-life clinical setting. The RETAIN study had certain limitations: (1) no central reading centre for CSFT measurement. (2) Re-treatment relied on VA loss, but the degree of loss was not defined and may thus vary by site, reflecting the clinical practice of the investigator. (3) Besides BCVA loss, changes in anatomical outcomes could also have been a consideration for re-treatment decisions. Examination of re-treatment visits in the PRN arm revealed that the VA loss from the previous visit was, on average, five letters and approximately 85% of re-treatments were accompanied by a loss in BCVA. In cases where treatment was administered without VA loss, the treatment decision may have been based on anatomical parameters, which may be a reflection of real-life clinical practice and is also consistent with the revised ranibizumab EU product label (2014)24. (4) Patients with previous stroke and transient ischaemic attack (high-risk patients) were excluded from the study. Finally, (5) the T&E regimen may have been too conservative, resulting in more injections than with PRN treatment, and its full potential with respect to reduction in number of visits and injections may still need to be determined.
In conclusion, the RETAIN study demonstrated that a ranibizumab T&E regimen is an appropriate alternative to a PRN regimen for management of DMO. This regimen allows for fewer clinic visits through extended intervals between treatments, thus providing the opportunity to reduce treatment burden and the potential to improve treatment compliance. Further studies using the T&E regimen with longer follow-up are required to explore the real-life implications using ranibizumab for treatment of patients with DMO. Reflecting this conclusion, the current ranibizumab EU product label (2014)24 allows intertreatment intervals to be extended per individual patient case based on the treating physician's opinion and assessment of disease activity.
Acknowledgments
The authors would like to thank Mohammed Najeeb Ashraf from Medical Operations (Global Business Services-Global Medical and Clinical Services, Novartis Healthcare, India) for his medical writing and editorial support and Stephanie Blick (Novartis Pharma AG, Basel, Switzerland) for her administrative, technical and logistic support towards the development of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
- Data supplement 3 - Online supplement
- Data supplement 4 - Online supplement
- Data supplement 5 - Online supplement
- Data supplement 6 - Online supplement
- Data supplement 7 - Online figures
- Data supplement 8 - Online table S1
- Data supplement 9 - Online table S2
- Data supplement 10 - Online table S3
Footnotes
Correction notice This article has been corrected since it was published Online First. ‘>300 μm’ was corrected to ‘<300 μm’ in the sentence “Patients were excluded if they showed structural damage within 0.5 disc diameter of the centre of the macula in the study eye likely to preclude improvement in VA following the resolution of macular oedema; BCVA >73 letters and central subfield thickness (CSFT) >300 μm in the study eye…”.
Collaborators Group members are listed in online supplementary file S1.
Contributors Conception and design of the study: CP, VB, SP and AW. Analysis and interpretation: CP, FF, SM, FR, KH, JS, VB, SP, WJS, AW and JF. Critical revision of the article: CP, FF, KH, FR, SM, WJS, AW, SP and JF. Final approval of the article: CP, FF, SM, FR, KH, JS, VB, SP, WJS, AW and JF. Data collection: VB, SP, WJS and AW.
Funding This work was sponsored by Novartis Pharma AG, Switzerland.
Competing interests Professor CP receives grants and fees from Alcon, Allergan, Bayer and Novartis outside of this work; FF reports personal fees from Novartis, personal fees from Bayer, personal fees from Allergan, outside the submitted work; SM receives grant and personal fee from Novartis, Bayer, Allergan and personal fee from Alcon; FR reports non-financial support and other from Novartis, during the conduct of the study; grants, personal fees and non-financial support from Novartis, outside the submitted work; JS reports personal fees from Novartis s.r.o., personal fees from Bayer s.r.o., personal fees from Ewopharma, spol. s r.o., outside the submitted work; VB, SP, WJS and AW report other from Novartis Pharma AG, during the conduct of the study; other from Novartis Pharma AG, outside the submitted work; JF reports grants from AIBILI, during the conduct of the study; personal fees from Allergan and Kemin, non-financial support from Alcon, Alimera and Bayer, outside the submitted work; KH reports fees for lectures and travel support from Bayer, Switzerland and Allergan, and fees for lectures from Novartis and Roche.
Patient consent Obtained.
Ethics approval The study adhered to the tenets of the Declaration of Helsinki, the International Conference on Harmonization and Good Clinical Practice guidelines. The protocol and amendments were approved by the Independent Ethics Committee or Institutional Review Board for each participating centre.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Full trial protocol can be assessed at: http://www.novctrd.com/ctrdWebApp/clinicaltrialrepository/displayFile.do?trialResult=11543
Linked Articles
- At a glance