
Abstract
Hyperglycaemia appears to be a critical factor in the aetiology of diabetic retinopathy and initiates downstream events including: basement membrane thickening, pericyte drop out and retinal capillary non-perfusion. More recently, focus has been directed to the molecular basis of the disease process in diabetic retinopathy. Of particular importance in the development and progression of diabetic retinopathy is the role of growth factors (eg vascular endothelial growth factor, placenta growth factor and pigment epithelium-derived factor) together with specific receptors and obligate components of the signal transduction pathway needed to support them. Despite these advances there are still a number of important questions that remain to be answered before we can confidently target pathological signals. How does hyperglycaemia regulate retinal vessels? Which growth factors are most important and at what stage of retinopathy do they operate? What is the preferred point in the growth factor signalling cascade for therapeutic intervention? Answers to these questions will provide the basis for new therapeutic interventions in a debilitating ocular condition.
Similar content being viewed by others

Main
Diabetic retinopathy, a major cause of blindness in developed countries, is characterised by hyperglycaemia, basement membrane thickening, pericyte loss, microaneurysms, IRMA and preretinal neovascularisation which can eventually lead to blindness through haemorrhage and tractional retinal detachment. Since it was first described in 19771 there has been much debate about the initiating factor(s) in diabetic retinopathy with lack of glucose control considered of major importance. This review aims to consider the role of hyperglycaemia in retinopathy and how this contributes to a change in the balance of regulators of the retinal vasculature.
Can glucose control in patients with diabetes prevent the onset of retinopathy?
The answer appears to be ‘yes’ from three hallmark trials undertaken on different continents: the Diabetes Control and Complications Trial (DCCT) undertaken in the USA in 1993,2 the United Kingdom Prospective Diabetes Study (UKPDS) in 19983 and a Japanese trial.4 DCCT highlighted that intensive glycaemic control can prevent or delay the development or progress of diabetic retinopathy by 76% in patients with type 1 diabetes within a primary prevention group over an average of 6.5 years. The UKPDS came to a similar conclusion when assessing glucose control and disease progression in patients with type 2 diabetes which supported the observations from the Japanese study.4 These trials demonstrated that blood glucose levels of diabetic patients at the first doctors visit indicated the outcome of their retinopathy and that glucose control early in the condition is probably vital to prevent or delay the onset of retinopathy.
How does hyperglycaemia regulate the progression of retinopathy?
Hyperglycaemia is associated with a variety of biological events identified in the progression of diabetic retinopathy (eg glucose transport, basement membrane thickening, pericyte loss, blood characteristics). Animal models such as the streptozotocin rat suggest that long-term hyperglycaemia is necessary to elicit changes to the retinal vasculature.5 Hyperglycaemia does not result in pathological changes in the retinal vasculature within the first 6 weeks. However, after this period proliferation of endothelial cells and swollen retinal vessels are observed. Interestingly, the retinal vessel lesions persisted even after the blood glucose levels have returned to normal. These abnormalities extended to include loss of pericytes and endothelial cells from the capillary beds and the appearance of microaneurysms. It is clear that there is a time point after which the progress of diabetic retinopathy is inevitable and reinforces that it is crucial to elicit preventative measures such as intensive blood glucose control at the very early stages of diabetes to prevent or slow progression of retinopathies. As will be discussed below, glucose can have a detrimental effect on a variety of biological processes.
Retinal endothelial cell glucose transport
To date, several possible mechanisms, including the polyol pathway,6 non-enzymatic glycation,7 oxidative stress8 and activation of protein kinase C (PKC)9 have been implicated in the development of retinopathy. These mechanisms are mostly dependent on excessive transport of glucose into retinal cells resulting in increased intracellular glucose levels. GLUT1, one of a family of glucose transporters, is exclusively responsible for glucose crossing the inner retinal-blood barrier.10,11 Surprisingly, the early stage of diabetic retinopathy exhibits a decrease in expression of GLUT1 in retinal endothelial cells, inferring that GLUT1 is not strongly linked with the development of retinopathy. Since expression of GLUT1 in the retinal pigment epithelium (RPE) is not affected by diabetes, it is likely that glucose entering the retina is greater across the RPE than across retinal endothelial cells.10 However, it has been proposed that an increase in density of relocalized GLUT1 in the inner blood-retinal barrier is enhanced by vascular endothelial growth factor (VEGF),12 a factor upregulated in retinopathy.
Retinal blood control
It is quite evident that there are numerous changes taking place in the vasculature that are associated with the early stages of diabetic retinopathy prior to the appearance of pathological changes. Furthermore, a duration of years or decades elapse before subtle changes lead to the observation of retinopathy. A typical example is the functional changes in the retinal vasculature13,14 resulting in a change of retinal tissue blood flow pattern including an increase in retinal blood flow and heterogeneity of distribution of retinal blood flow.15 The retinal vasculature relies on local mechanisms to regulate appropriate blood flow.16 To date at least 10 regulating factors have been proposed for control of blood flow.17 They fall into two main classes: endothelium-derived relaxing factors (nitric oxide, prostacyclin, endothelium-derived hyperpolarizing factor) and endothelium-derived contracting factors (endothelin, cycloxygenase products) which inhibit or stimulate respectively the underlying smooth muscle cells and pericytes. Importantly, many of these factors are regulated by local glucose levels.
Endothelium-derived relaxing factors
Nitric oxide
Nitric oxide (NO) acts to maintain appropriate arteriolar vasodilation as well as stablilizing platelets.18,19,20,21,22,23,24,25 It is synthesized in cells from L-arginine to L-citrulline via activation of a calcium-dependent nitric oxide synthase. In response to platelet-derived products, hormones and mechanical changes such as transmural pressure,26 endothelial cells release NO into the surrounding milieu. NO enters smooth muscle cells and activates soluble guanylate cyclase. This results in increased cyclic guanosine 3′,5′-monophosphate (cGMP), which is responsible for subsequent relaxation of the smooth muscle cells through a decrease in Ca2+ and dephosphorylation of myosin light chains.27 In the retinal vascular bed there is a constant basal release of NO which maintains the retinal circulation in a constant state of vasodilation.
Evidence indicates that there are at least three mechanisms for diminishing production and/or increasing quenching of NO by hyperglycaemia. First, hyperglycaemia causes de novo synthesis of diacylglycerol (DAG), leading to activation of PKC. The consequence of PKC activation is that PKC reduces the capacity of a number of agonists to increase intracellular Ca2+ and stimulate NO synthesis.28,29,30 Furthermore, PKC may provoke expression of superoxide in endothelial cells which quenches NO.31 Second, hyperglycaemia activates the polyol pathway by increasing substrate-glucose for endothelial aldose reductase.32 This enzyme converts glucose to sorbitol by a reaction that oxidizes NADPH and reduces its availability (NADPH is one of the cofactors for NO synthesis).33 Third, hyperglycaemia generates non-enzymatic glycated proteins34 which lead to subsequent superoxide generation resulting in inactivation of NO. Additionally, glycated proteins can directly quench NO.35
Prostacyclin (PGI2)
PGI2 is generated from the metabolism of arachidonic acid via cyclooxygenase36 and is complementary to NO. PGI2 stimulates enhanced production of cyclic adenosine 3′,5′-monophspate (cAMP) through adenylcyclase in smooth muscle cells and pericytes, leading to relaxation of those cells.37 In endothelial cells PG12 synthesis is mediated by prostacylin-stimulating factor (PSF), which is constitutively expressed in retinal pericytes.38 However, early hyperglycaemia results in a transient decrease in PSF production, causing a decrease in PGI2 synthesis.39 In animal models it has been shown that after the induction of diabetes an early decrease of PSF in the retina is followed by an increase in PSF levels. Hyperglycaemia also inhibits PGI2 synthesis through generating lipid peroxide and arachidonic acid via microsomal desaturase.39 Intriguingly, normal levels or even increased of PGI2 in diabetes have been reported.39 This might be due to upregulation of phospholipase A2 (PLA2) providing increased substrate for PGI2 synthesis.
Endothelium-derived hyperpolarizing factor (EDHF)
EDHFs are substances that are distinct from nitric oxide (NO) or prostacyclin (PGI2). EDHFs are thought to mediate endothelium-dependent hyperpolarization of vascular smooth muscle cells or pericytes. EDHF contributes greatly to vascular control of small-diameter vessels and microvessels involved in the local regulation of peripheral vascular resistance and thus in the distribution of blood flow.40 By contrast, NO and PGI2 are more committed to regulation of large-diameter vessels. A list of potential agents/cellular events which could function as EDHF include eoxyeicosatrienoic acid (EET),41,42,43,44,45,46 hydrogen peroxide,47 potassium efflux,48 and gap junction communication between endothelial cells and smooth muscle cells.49 For example, EET is released from endothelial cells in response to acetylcholine (ACh) and hyperpolarizes smooth muscle cells by opening Ca2+-activated K+ channels and causing vasodilatation.47 A line of evidence for a role of EDHF in diabetic retinal vascular dysfunction is inferred from data indicating that in diabetes, endothelium-dependent hyperpolarizations are diminished by hyperglycaemia largely due to a defective vascular response to EDHF.50
Endothelium-derived contracting factors
Endothelin-1 (ET-1)
ET-1 is a powerful vasoconstrictor peptide. The circulation levels of ET-1 are low under normal conditions suggesting that ET-1 acts as a local regulatory factor.51 ET-1 causes vasodilation at low concentrations and a constrictive response at high concentrations via the interaction with endothelin receptors (ETA-, and ETB) on smooth muscle cells and pericytes.52,53,54 ET-1-induced activation of endothelin receptors, linked to voltage-operated Ca2+ channels, either opens the gates of the Ca2+ channel leading to influx of Ca2+,55 or induces activation of phospholipase C (PLC) by the formation of diacylglycerol (DAG), shifting Ca2+ from intracellular stores and increasing intracellular Ca2+.56,57 Increased intracellular Ca2+ in smooth muscle cells in turn induces lasting contractile effects. Hyperglycaemia is most likely to induce an increase in ET-1 levels in retinal vascular cells.58 PKC and mitogen-activated protein kinase (MAPK) are activated in retinal microvascular cells by the elevation of glucose levels.59,60 Thus, PKC and MAPK pathways enhance the ET-1 transcription rate.
Cyclooxygenase products
Cyclooxygenase products induce vasoconstriction and include thromboxane (TX),61 prostaglandin (PG)62 and lipid peroxides (LPO)63 which can be found in endothelial cells and platelets. However, overproduction of these factors has been detected in diabetic retinopathy. For instance, PKC, enhanced by hyperglycaemia, activates phospholipase A2 (PLA2) which sequesters arachidonic acid from membrane phospholipids.64 Arachidonic acid is a substrate that is catalyzed by cyclooxygenase and lipoxygenase promoting LPO generation.65
Retinal capillary cell death
Histological analyses of diabetic retina demonstrate localised regions of non-perfused acellular ‘vessels’ consisting solely of basement membrane.66 The early and progressive loss of retinal capillary cells, including pericytes and endothelial cells, inevitably leads to microaneurysms and vascular obstruction. Retinal capillary cell death unquestionably has a major impact on retinal vessels in diabetes and in the case of pericyte loss occurs long before the onset of proliferative diabetic retinopathy (PDR). However, capillary cell death, specifically pericytes, has been found to be rare or absent from capillaries of the optic nerve and cerebrum.67 Perhaps this is evidence that disappearance of retinal capillary cells may be due to a local disorder rather than systemic abnormalities such as hyperglycaemia? However, hyperglycaemia has been shown to induce pericyte apoptosis both in vivo and in vitro,67,68 with in vitro evidence that cell death is exacerbated when glucose levels fluctuate between hyper- and normoglycaemia as often occurs in poorly controlled diabetes.68
Polyol pathway
There is limited evidence that hyperglycaemia can, in tissues such as the retina that do not require insulin for cellular glucose uptake, induce polyol pathway hyperactivity and aldose reductase expression.69 In addition to its well-documented glucose metabolism role, the polyol pathway has been found to cause a loss of retinal capillary cells and that this involves aldose reductase, a rate-limiting enzyme of the polyol pathway that reduces glucose to sorbitol.70,71 Aldose reductase inhibitor has been reported to inhibit the high glucose-induced death of retinal capillary cells.72,73 Sorbitol, a common organic osmolyte in many cells, accumulates in retinal capillary cells in response to hyperglycaemia and causes hyperosmolality of the cells.74,75 Thus, hyperosmolality induces an increase in intracellular water and lactate production, and a decrease in oxygen uptake. The other part of the polyol pathway involves glutathione reductase reducing NADPH to NAD, in which aldose reductase competes with NADPH.76,77 NADPH is required not only by glutathione reductase for the reduction of oxidized glutathione (GSSG) to glutathione (GSH), but also by aldose reductase for conversion of glucose to sorbitol. Reduced NADPH may also be responsible for dysfunction of endothelial enzymes, for example eNOS.33 In addition, hyperactivity of the polyol pathway requires large quantities of ATP78 and may consume the energy required for production of endothelium-dependent relaxation factors.
Glycation pathway
During normal ageing, glucose binds non-enzymatically to free amino groups in proteins and forms Amadori adducts through a series of oxidative and non-oxidative reactions.79 Hyperglycaemia and oxidative stress probably confer on Amadori adducts the opportunity to continue to rearrange and generate irreversible advanced glycation end products (AGEs) in diabetes.80 The impact of AGEs on retinal capillary cells is related to their capacity to accumulate in tissues over time, to form cross-links and to generate oxygen-derived free radicals.81,82,83,84 Additionally, binding of AGEs with their receptors may provoke sustained cell activation and further oxidative stress.
Oxidative stress
Oxidative stress is defined as an increase in the steady-state levels of reactive oxygen species. There are several endogenous enzyme systems that protect the cell and tissue from oxidative stress, for example superoxide dismutase (SOD),85 catalase,86 and glutathione peroxidase (GSH-Px).87 Although there is controversy about the antioxidant status in diabetes, several studies report decreased levels of SOD and GSH-Px in both clinical and experimental diabetes,83,84,88,89 indicating an impaired defence system for free radical scavenging. Sources of reactive oxygen species in diabetes may include autoxidation of glucose, AGE-formation and the binding of AGE to AGE receptors, increased substrate flux through the polyol pathway and stimulation of eicosanoid metabolism. 8-epi-PGF(2alpha), one of the prostaglandin-F(2)(PGF(2))-like compounds produced during peroxidation of arachidonic acid (AA) by a mechanism independent of the cyclo-oxygenase,90 has recently been detected in the retina during diabetes. This provides direct evidence that oxygen-derived radicals produced during prostanoid synthesis rather than the prostanoids themselves are responsible for endothelial dysfunction in diabetes mellitus.91 Oxygen-derived free radicals may impair endothelium-dependent vasodilation through inactivation of NO.92 In addition, oxidative stress can cause an increase in the conversion of deoxyguanosine to 8-oxo, 2′-deoxyguanosine in DNA.93 Both the altered gene profile of scavenging enzymes94 and overexpression of the cell death protease gene95 are believed to increase apoptosis of retinal capillary cells in diabetic retinopathy.
What causes retinal ischaemia?
Retinal ischaemia is generally believed to result from structural and functional derangement of the retinal microcirculation. The formation of acellular capillaries is a major histological feature of the ischaemic retina.96,97 The capillary basement membrane tubes without endothelial cells and pericyte nuclei firstly only occur singly or as small groups scattered about the retinal. Later they are found in large clusters with atrophic arterioles.98,99
Several possible mechanisms have been proposed for the appearance of retinal ischaemia in diabetes. These include thickened basement membranes, platelet aggregation, leukocyte activation/adherence or a combination thereof. Furthermore, hyperglycaemia is likely to be a major risk factor.
Retinal basement membrane thickening
In diabetes, early hyperglycaemia is sufficient to increase the synthesis of basement membrane components in the retina100 which in turn may contribute to the closure of capillaries. For example, mRNA for fibronectin and collagen types I, III, IV(α1, α2), and V are found to be upregulated in the retinal basement membrane of diabetic retinopathy. Furthermore, in the retina of diabetic patients increased immunostaining is observed compared to normals for vitronectin in the arterioles,101 collagen types I, II, III, IV in the venules102 and laminin and fibronectin in both arterioles and venules.103 Animal models also show that retinal expression of collagen type IV and fibronectin increases in hyperglycaemic rats.104,105,106 Diabetic basement membrane thickening appears to involve qualitative alterations of specific basement membrane markers at an advanced disease stage, with the appearance of diabetic retinopathy. For instance, abnormal accumulation of several extracellular matrix components in retinal basement membranes may trigger the deposition of small tenascin-C isoforms in the blood vessel walls.107 The expression of tenascin,108 an extracellular matrix glycoprotein, originally found to modulate organogenesis in tendinous and glial tissue, suggests that this glycoprotein may promote retinal basement membrane thickening.
Platelet aggregation
Diabetic retinopathy is associated with an increased number and size of platelet-fibrin thrombi in the retinal capillaries compared to normal.109 These thrombi can contribute to capillary obliteration and retinal ischemia. It has been reported that chronic hyperglycemia causes an increase in diacylglycerol (DAG) levels in the retina, which may activate PKC.110 Through increased intracellular Ca2+, PKC stimulates endothelial cells, leuckocytes and platelets to produce platelet-activating factor (PAF).111,112,113 PAF, confined to membranes, stimulates PAF receptors114 on platelets, inducing activation of these platelets. Activated platelets produce a number of platelet-derived microparticles,115,116 which contribute to thrombus formation by providing and expanding a catalytic surface for the coagulation cascade. Pathological levels of fluid shear stress in abnormal retinal blood vessels affected by hyperglycaemia may cause both further platelet aggregation and shedding of more microparticles from the platelet plasma membrane.117 In addition, elevated sorbitol in the retina and erythrocytes can reduce vascular prostacyclin accompanied by an increased synthesis of thromboxane via induction of adenosine diphosphate (ADP)118 or collagen119 in whole blood. The imbalance of thromboxane and prostacylin enhances platelet hyperactivity.120 Adhesion proteins121 that are cofactors in the aggregation of human platelets and mediating the adenosine diphosphate (ADP)-induced response of these cells are also increased significantly.
Leukocyte activation and adherence
Although diabetic retinopathy generally is not considered as an inflammatory disease, leukocytes adhere to the retinal vascular endothelium early in experimental diabetic retinopathy.122 Excess activation of endothelial PKC promotes PAF synthesis in diabetes.110,123,124 PAF stimulates PAF receptors on peripheral leukocytes rolling on the lumenal endothelial membrane leading to their activation.125 Activated leukocytes also synthesise PAF126 and leukotriene B4 (LTB4),127,128 which further enhance activation of leukocytes via autocrine action. β2 integrins129,130 on activated leukocytes enable the leukocytes to adhere tightly to the endothelial cell via binding intercellular adhesion molecule-1 (ICAM-1).131 ICAM-1 is upregulated by endothelial PKC which normally acts to stabilize mRNA of β2 integrins.132,133 Physiologically, nitric oxide (NO) plays a role in modulating leukocyte activation and adherence. Presumably, NO deficiency can allow leuckocytes to escape from NO control, leading to leukocyte activation and adherence.134,135 Furthermore, leukocytes in diabetes have been reported to be less deformable due to actin polymerization and increase in their viscosity.136 Alteration in retinal blood flow could reduce pressure gradients across retinal capillaries owing to stenotic or constricted arterioles resulting in activated leukocytes becoming wedged in capillaries and postcapillaries and obstructing retinal microvessels.137,138
What is the importance of retinal hypoxia?
The retinal vasculature is relatively sparse in order to minimise optical interference in the light path. This results in a large oxygen tension difference between retinal arteries and veins which can easily be compromised if damage occurs to the vascular bed. Capillary nonperfusion, loss of retinal capillaries, AGEs and/or oxidative stress can lead to progressive retinal hypoxia.139,140,141,142,143
Acute hypoxia rapidly activates retinal vascular endothelial cells to release inflammatory cytokines.144 These inflammatory mediators are able to recruit and promote the activation and adherence of leukocytes,145 which contribute to the obstruction of retinal capillaries, leading to further hypoxia. Chronic hypoxia is, at least in the retina, sufficient to induce the expression of angiogenic growth factors,146,147 resulting in the characteristic retinal neovascularization associated with proliferative diabetic retinopathy (PDR). The observation that retinal neovascularization occurs adjacent to the nonperfused area148,149 supports the hypothesis that angiogenic factors are released from hypoxic tissue. In the majority of instances regression of preretinal new vessels can be achieved through the use of scatter laser photocoagulation.150,151 While the mechanism of action of scatter laser photocoagulation remains elusive, there is support for the hypothesis that destruction of retinal tissue makes more oxygen available for the retina and returns it to normoxia.152
The whole picture of how hypoxia induces and increases the expression of angiogenic factors is not clear, but parts of the puzzle are beginning to emerge. A cytosolic flavoheme protein acts as an oxygen sensor that detects decreased oxygen tension and activates transcription factors through signal transduction pathways.153,154 Hypoxia inducible factor 1 (HIF-1) is a major transcription factor.155 The activation of HIF-1 depends upon signaling-dependent rescue of its alpha-subunit from oxygen-dependent degradation in the proteasome and formation of a heterodimer with HIF-1beta, which then translocates to the nucleus and impacts on the transcription of genes that are upregulated by hypoxia156,157,158 Activation of HIF-1 has been shown to increase production of a variety of factors implicated in the pathogenesis of diabetic retinopathy (eg ischaemic retina159).
How do growth factors play a pivotal role in diabetic retinopathy?
It is now quite evident that there is a plethora of growth factors which regulate the retinal vasculature and are involved in the development and progression of diabetic retinopathy. However, identifying the role of each growth factor is difficult since growth factors can act alone or, as appears to be more often the case, interact with each other. Examples include: one growth factor inducing the synthesis of a more potent growth factor, synergy between growth factors and commonality in the downstream transduction cascade. In addition, knockout studies have shown that if the action of a growth factor is negated other growth factors are synthesised to overcome this deficit.
The last three decades have seen the discovery of a large number of growth factors, most of which have been implicated to a greater or lesser extent in diabetic retinopathy. Of these vascular endothelial growth factor has received considerable attention of late due to its potent angiogenic activity.160
Vascular endothelial growth factor (VEGF)
VEGF is a potent angiogenic factor capable of stimulating endothelial cells to degrade extracellular matrix, migrate, proliferate and form tubes.161,162,163 Recently, it also has been found to act as a survival factor for newly formed vessels.164 VEGF exerts its functions on endothelial cells via interaction with cellular receptors Flt-1 (VEGFR-1) and Flk-1/KDR (VEGFR-2),165,166,167,168,169,170,171 both receptor tyrosine kinases. Interaction between VEGF and its receptors initiates a signal transduction pathway, and thus induces phosphorylation of proteins downstream in endothelial cells, including phospholipase Cγ (PLCγ),172,173 phosphatidylinositol 3-kinase (PI3-Kinase)174,175 and guanine 5′ triphosphate (GTP)ase-activating protein.176 Phosphorylation of PLCγ may be more important for retinal endothelial cells. Phosphorylated PLCγ converts inositol phosphate into diacylglycerol (DAG), causing activation and translocation of PKC that engages subsequent changes in endothelial cells.177,178 It is generally considered that activation of the Flt-1 receptor regulates the metabolism of a range of vascular and non-vascular cells while KDR which is relatively specific for vascular endothelial cells promotes migration and proliferation.
Increased levels of VEGF have been identified in the vitreous and the retina of patients with diabetes.162,179,180,181 This increase is likely to be hypoxia-induced since elevated levels of VEGF protein and mRNA are present in the ischaemic retina adjacent to the areas of neovascularization in diabetic animals and human pathology specimens.182,183,184 Moreover, in vitro hypoxia also induces expression of VEGF mRNA in retinal cells.185 Hypoxia is also reported to induce expression of VEGF receptors in endothelial cells indicating that sensitivity to VEGF is enhanced in the ischaemic retina.186,187
VEGF also appears to play an early role in the development of diabetic retinopathy. VEGF is clearly elevated in diabetic retinal tissue without overt retinopathy and is likely to initiate the increased permeability associated with the retinal vasculature in diabetes.186,188,189,190 Animal experiments have convincingly produced clinical features of nonproliferative diabetic retinopathy by repeated intravitreal injections of VEGF.191 VEGF has long been known to increase the permeability of vascular endothelium, which may involve rearrangement of interendothelial junctional proteins,192,193 including VE-cadherin, tight junction proteins (eg occludin and zonula occluden 1) in retinal endothelium. Such effects presumably underlie the increasd risks of vessel leakage and macular edema in diabetic retinopathy. Clinical experience suggests that there is fluctuation of retinal blood flow in patients with diabetes, who have a decrease in retinal blood flow at the early stage of retinopathy, with a progressive increase in retinal blood flow in more advanced stages.190,194 Similar changes in retinal blood flow are observed in diabetic animals following intravitreal injections of VEGF.191 Such observations support the concept that VEGF not only contributes to retinal neovasculariztion, but also produces earlier changes in diabetic retinopathy. Interestingly, inhibition of VEGF activity by specific antisense oligonucleotides,195 VEGF-neutralizing antibodies196 or soluble receptors197 is insufficient to completely prevent neovascularization. The incomplete inhibition of neovascularization indicates that retinal neovascularization may be driven at least in part by alternative angiogenic factors.
Alternative angiogenic factors
A plethora of other angiogenic factors including insulin-like growth factor-I (IGF-I), basic fibroblast growth factors (bFGF or FGF2), platelet-derived growth factor (PDGF), hepatocyte growth factor/scatter factor (HGF/SF), placenta growth factor (PIGF) and angiopoietin2 (Ang2) have been implicated in retinal neovascularization.
IGF-I
A role for a pituitary associated factor was hypothesised over 40 years ago when retinal neovascularisation was found to regress after pituitary infarction.198 Subsequently the pituitary factor has been identified as growth hormone and the mitogenic mediator of growth hormone action is Insulin-like growth factor-I (IGF-I).199 IGF-I was one of the first growth factors to be directly linked with diabetic retinopathy.200 Initial reports demonstrated an acute increase in serum levels of IGF-I preceded the onset of proliferative diabetic retinopathy (PDR) in animal models.201,202 Subsequently, increased IGF-I levels were measured in the vitreous of patients with PDR,203 indicating that IGF-I may play a role in retinal neovascularization and that the localised effect of IGF-I may be more important than its systemic role in the development of neovascularization. It has been proposed that leakage across the blood–retina barrier and high serum levels of IGF might be the major source for vitreous IGF levels. Confirmation has come from in vitro studies204,205,206 showing that IGF-I can induce almost all steps of the angiogenesis process including endothelial cell proliferation, migration and basement membrane degradation. In vivo, retinal angiogenesis has been confirmed following application of IGF-I to the retina of rabbits.207 IGF-I exerts its effect on endothelial cells via coupling with the IGF-I receptor (IGF-IR). Two pathways are prominent in IGF-I signalling, the Ras/Raf/MAPK cascade208 and the PI 3-kinase system,209 both of which promote cell survival and proliferation.210,211 IGF-I is regulated by a family of insulin-like growth factor binding proteins (IGFBPs)212 which can inhibit or potentiate IGF-I activity depending on a number of parameters such as their affinity for IGF-I, the biological system in question and post-translational modifications.213 IGFBPs 1, 2 and 3 have been reported to be significantly increased in vitreous from patients with PDR but not in non-ischaemic eye disease.214,215,216 The contribution of leakage of the blood retinal barrier and local synthesis to vitreous levels of these proteins is unclear, however, local synthesis will certainly contribute. Comparison of cultured retinal endothelial cells from normal and diabetic donors demonstrates a decrease in IGF-1 and an increase in the IGFBP 1, 2 and 5 message for diabetic cultures compared to normals.217 An important link between IGF-1 and IGFBP expression and diabetic retinopathy is hypoxia. Several studies in vitro have shown that IGF-I and IGFBPs are subject to regulation by hypoxia.218,219,220
bFGF
Whether basic fibroblast growth factor (bFGF) participates in the stimulation of retinal neovascularization has been a matter of considerable controversy. bFGF is stored at high concentration within the extracellular matrix (ECM) as an inactive complex, and released when endothelial cells dissolve ECM via the release of proteases.221,222,223 bFGF and hypoxia act synergistically to not only induce mitogenesis in endothelial cells, but also to upregulate VEGF in smooth muscle cells and endothelial cells, resulting in retinal angiogenesis.224 However, the fact that bFGF-deficient animal models develop the same degree of retinal neovascularization as wild-type animals argues against a major angiogenic role for bFGF in diabetic retinopathy.225 Although bFGF may not directly induce retinal neovascularization, it can regulate VEGF expression in retinal vascular cells.226
PDGF
The platelet-derived growth factor (PDGF) family comprises three isoforms, PDGF AA, BB and AB, which act via PDGF receptor subunits (α- and β-). PDGF is widely expressed upon tissue injury and repair227,228 and the PDGF BB isoform is induced by hypoxia.229,230 Additionally, significantly elevated concentrations of PDGF AB are found in the vitreous and preretinal membranes of patients with proliferative diabetic retinopathy (PDR).231 PDGF may act directly on endothelial cells232 that are engaged in angiogenesis or that express PDGF receptor beta-subunits.233,234 However, it is reported that PDGF AB is also elevated in ischemic non-diabetic retinopathy, indicating that ischemia rather than diabetes per se might be a strong stimulator of PDGF production in the retina.231 Since PDGF is known to induce the generation of a vascularized connective tissue stroma in many angiogenic and proliferative processes,235 retinal neovascularization in response to PDGF-BB may be partially due to its direct effects on the formation of fibrovascular retinal membranes.
HGF/SF
Hepatocyte growth factor/scatter factor (HGF/SF)236 is a most potent mitogenic factor for a number of cell types, including hepatocytes,237 myeloid precursor cells,238 and various epithelial239 and endothelial cells.240 HGF/SF also promotes epithelial and endothelial cell motility in addition to regulating tube morphogenesis and tube branching.241,242 HGF/SF binds to the c-Met receptor243 and initiates signaling via activation of both protein kinase C (PKC)244 and phosphatidylinositol 3-kinase (PI3-Kinase),245,246 inducing MAPK phosphorylation that is critical for migration and growth. HGF/SF and its receptor levels have been shown to significantly increase in the vitreous of diabetic patients compared to normal control groups.247 HGF/SF can also induce VEGF production by a variety of cells and tissues.248,249,250 Since VEGF does not appear to mediate these initial HGF effects it suggests that HGF/SF acts as a co-factor promoting retinal neovascularization.
PIGF
PIGF is a member of the VEGF family and shares 35% primary sequence homology with VEGF.251 However, unlike VEGF which binds to both VEGFR-1 and VEGFR-2, PIGF binds only to VEGFR-1.252 Furthermore, PIGF can form a heterodimer with VEGF presumably regulating VEGF-receptor binding through both VEGFR-1 and VEGFR-2. The different affinity for VEGFRs has been shown to influence endothelial cell behaviour. Generally, the ligand-receptor interaction is reflected in the different signal transduction pathways that involve different signal components. For example, as a marker for DNA synthesis, mitogen-activated protein kinase (MAP kinase) is activated in endothelial cells stimulated both by VEGF and PIGF,253 whereas phospholipase C-γ (PLC-γ) which plays a role in cell migration is only tyrosine phosphorylated by VEGF stimulated cells.254 In vitro studies indicate that VEGF essentially has no effect on Flt-1 expressing cells while PIGF can induce DNA synthesis in Flt-1 expressing cells, leading to mitogenesis but not migration. However, PIGF appears to be different in vivo and not only stimulates proliferation of endothelial cells, but also induces angiogenesis.255 Furthermore, the PIGF/VEGF heterodimer induces angiogenesis more effectively than the PIGF homodimer alone.256
There is considerable evidence to support a role for PIGF in diabetic retinopathy. PIGF levels are upregulated in the vitreous of patients with PDR257,258 and PIGF protein localises to areas of active retinal neovascularisation.258 Interestingly PIGF was found to induce secretion of VEGF and was co-expressed with VEGF.257 The PIGF gene has also been shown to be elevated in PDR retinas259 and in retinas in a mouse model for retinopathy of prematurity.260 While the origin of PIGF in vivo remains to be determined, cell culture studies demonstrate that both endothelial cells and pericytes express PIGF mRNA.261 PIGF was more highly expressed in endothelial cells compared to pericytes. It is likely that in PDR PIGF potentiates the effect of VEGF either via enhancing expression of VEGF or the formation of a heterodimer with VEGF.255
Angiopoietin
Another family of tyrosine kinase receptors which play an important role in angiogenesis are the Tie (tyrosine kinase with immunoglobulin and epidermal growth factor homology domains) receptors.262 To date two receptors have been identified, Tie1 and Tie2. No ligand has been identified for Tie1 while the best characterized ligands for Tie2 are angiopoietin1 (Ang1)263 and angiopoietin2 (Ang2),264 both sharing 60% amino acid homology. Ang1 and Ang2 appear to have different effects on endothelial cells. Ang1 induces tyrosine phosphorylation of Tie2 and activates the downstream signalling pathway to promote vascular maturation,265 whereas Ang2 acts as a naturally occurring antagonist of Ang1 by competing for binding to Tie2 and blocking Ang1 induced Tie2 phosphorylation.264 Ang1 has been reported to induce sprouting and chemotaxis in endothelial cells in vitro,266,267 whereas Ang2 appears to play a critical role in vascular remodelling.268,269 It has been shown that Ang2 is upregulated during angiogenesis in retinal development270 and in mouse models of ischaemia-induced retinal neovascularisation.270,271 A recent study has determined the spatial and temporal expression of Ang1, Ang2 and the Tie2 receptor during the pathogenesis of diabetic retinopathy (Smith, personal communication). Interestingly, Ang1 protein was upregulated in PDR while Ang2 was downregulated suggesting that while angiopoietins play a key role in the pathogenesis of diabetic retinopathy their action may differ depending on the development stage of the vasculature and the type of disease.
Pigment epithelium-derived factor
Pigment epithelium-derived factor (PEDF) is a 50-kDa glycoprotein originally identified in RPE cells.272 Subsequently, PEDF mRNA has been found in most cell types and appears to have a wide variety of functions, eg neural development, neuronal protection and as a regulator of neovascularisation.272,273
PEDF is seen to be downregulated in eyes with active PDR274 suggesting that PEDF is a negative regulator of angiogenesis. Support for this comes from studies which have shown that: (a) PEDF regulates the development of the retinal vasculature;275 (b) is downregulated by hypoxia; and (c) PEDF inhibits retinal and choroidal neovascularisation in a number of animal models.276,277 That the latter was achieved by adenoviral transfection opens the possibility for gene therapy to upregulate PEDF and inhibit abherrant angiogenesis. It is unclear how PEDF exerts its effect but in vitro experiments show that PEDF may inhibit neovascularisation by promoting apoptosis of endothelial cells.277 The mode of action of PEDF will be clarified once its receptor(s) and downstream transduction pathway(s) have been identified.
Concluding comments
Research over the past few decades has provided ample evidence that hyperglycaemia is one of the main forces driving the onset and progression of diabetic retinopathy. Several mechanisms, by which hyperglyaemia causes retinal capillary damage include increased polyol pathway, activation of protein kinase C, increased non-enzymatic glycation and generation of reactive oxygen species (Figure 1). Furthermore, hyperglycaemia-induced events regulate the synthesis of a variety of growth factors implicated in retinopathy. A number of key growth factors have emerged of which the VEGF and PEDF families are critically important. The question we now ask is can the therapeutic modulation of growth factor pathways prove efficacious in the intervention in diabetic retinopathy at clinic level?

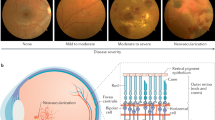
Schematic diagram of the pathogenesis of diabetic retinopathy. Abbreviations: NO, nitric oxide; PGI2, prostacyclin; VEGF, vascular endothelial growth factor; TGFβ, transforming growth factor beta; AGEs, advanced glycation endproducts; PIGF, placenta growth factor; PEDF, pigment epithelium-derived factor.
References
Cohen MP, Jasti K, Rye DL . Somatomedin in insulin-dependent diabetes mellitus. J Clin Endocrinol Metab 1977; 45: 236–239
The DCCT Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Eng J Med 1993; 329: 977–986
UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352: 837–853
Ohkubo Y, Kishikawa H, Araki E, Miyata T, Isami S, Motoyoshi S et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract 1995; 28: 103–117
Alder VA, Su EN, Yu DY, Cringle SJ, Yu PK . Diabetic retinopathy: early functional changes. Clin Exp Pharmol Physiol 1997; 24: 785–788
Yoshii H, Uchino H, Ohmura C, Watanabe K, Tanaka Y, Kawamori R . Clinical usefulness of measuring urinary polyol excretion by gas-chromatography/mass-spectrometry in type 2 diabetes to assess polyol pathway activity. Diabetes Res Clin Pract 2001; 51: 115–123
Chibber R, Molinatti PA, Kohner EM . Intracellular protein glycation in cultured retinal capillary pericytes and endothelial cells exposed to high-glucose concentration. Cell Mol Biol 1999; 45: 47–57
Gurler B, Vural H, Yilmaz N, Oguz H, Satici A, Aksoy N . The role of oxidative stress in diabetic retinopathy. Eye 2000; 14: 730–735
Ways DK, Sheetz MJ . The role of protein kinase C in the development of the complications of diabetes. Vitam Horm 2001; 60: 149–159
Badr GA, Tang J, Ismail-Beigi F, Kern TS . Diabetes downregulates GLUTI expression in the retina and its microvessels but not in the cerebral cortex or its microvessels. Diabetes 2000; 49: 1016–1021
Ban Y, Rizzolo LJ . Regulation of glucose transporters during development of the retinal pigment epithelium. Brain Res Dev Brain Res 2000; 121: 89–95
Sone H, Deo BK, Kumagai AK . Enhancement of glucose transport by vascular endothelial growth factor in retinal endothelial cells. Invest Ophthalmol Vis Sci 2000; 41: 1876–1884
de Abreu JR, Silva R, Cunha-Vaz JG . The blood-reinal barrier in diabetes during puberty. Arch Ophthalmol 1994; 112: 1338–1348
Cunha-Vaz JG, Leite E, Sousa JC, de Abreu JR . Blood-retinal barrier permeability and its relation to progression of retinopathy in patients with type 2 diabetes. A four-year follow-up study. Graefes Arch Clin Exp Ophthalmol 1993; 231: 141–145
Alder VA, Su EN, Yu DY, Cringle S, Yu PY . Overview of studies on metabolic and vascular regulatory changes in early diabetic retinopathy. Aust NZJ Ophthalmol 1998; 26: 141–148
Laties AM . Central retinal artery innervation. Absence of adrenergic innervation to the intraocular branches. Arch Ophthalmol 1967; 77: 405–409
Chen YF, Oparil S . Endothelial dysfunction in the pulmonary vascular bed. Am J Med Sci 2000; 320: 223–232
Vallance P, Collier J, Moncada S . Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet 1989; 2: 997–1000
Pohl U, Wagner K, de Wit C . Endothelium-derived nitric oxide in the control of tissue perfusion and oxygen supply: physiological and pathophysiological implications. Eur Heart J 1993; 14 (Suppl 1): 93–98
Stamler JS, Loh E, Roddy MA, Currie KE, Creager MA . Nitric oxide regulates basal systemic and pulmonary vascular resistance in healthy humans. Circulation 1994; 89: 2035–2040
Radomski MW, Palmer RM, Moncada S . Comparative pharmacology of endothelium-derived relaxing factor, nitric oxide and prostacyclin in platelets. Br J Pharmacol 1987; 92: 181–187
Arndt H, Russell JB, Kurose I, Kubes P, Granger DN . Mediators of leukocyte adhesion in rat mesenteric venules elicited by inhibition of nitric oxide synthesis. Gastroenterology 1993; 105: 675–680
Kubes P, Suzuki M, Granger DN . Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A 199; 88: 4651–4655
Bath PMW, Hassall DG, Gladwin AM, Palmer RM, Martin JF . Nitric oxide and prostacyclin. Divergence of inhibitory effects on monocyte chemotaxis and adhesion to endothelium in vitro. Arterioscler Thromb 1991; 11: 254–260
Heller R, Bussolino F, Ghigo D, Garbarino G, Pescarmona G, Till U, Bosia A . Nitrovasodilators inhibit thrombin-induced platelet-activating factor synthesis in human endothelial cells. Biochem Pharmacol 1992; 44: 223–229
Karmakar N . Interaction of transmural pressure and shear stress in the transport of albumin across the rabbit aortic wall. Atherosclerosis 2001; 156: 321–327
Imai T, Morita T, Shindo T, Nagai R, Yazaki Y, Kurihara H et al. Vascular smooth muscle cell-directed overexpression of heme oxygenase-1 elevates blood pressure through attenuation of nitric oxide-induced vasodilation in mice. Circ Res 2001; 89: 55–62
Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL . Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A 1992; 89: 11059–11063
Inoguchi T, Xia P, Kunisaki M, Higashi S, Feener EP, King GL . Insulin’s effect on protein kinase C and diacylglycerol induced by diabetes and glucose in vascular tissues. Am J Physiol 1994; 267: E369–E379
Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science 1996; 272: 728–731
Giugliano D, Ceriello A, Paolisso G . Oxidative stress and diabetic vascular complications. Diabetes Care 1996; 19: 257–267
Dunlop M . Aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney Int 2000; 58 (Suppl 77): S3–S12
Stevens MJ, Dananberg J, Feldman EL, Lattimer SA, Kamijo M, Thomas TP et al. The linked roles of nitric oxide, aldose reductase and (Na+, K+)-ATPase in the slowing of nerve conduction in the streptozotocin diabetic rat. J Clin Invest 1994; 94: 853–859
Ceriello A . Hyperglycaemia: the bridge between non-enzymatic glycation and oxidative stress in the pathogenesis of diabetic complications. Diabetes Nutr Metab 1999; 12: 42–46
Bucala R, Tracey KJ, Cerami A . Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J Clin Invest 1991; 87: 432–438
Levine L . Lactacystin stimulates arachidonic acid metabolism in rat liver cells: effects of cell density on arachidonic acid release, PGI2 production and cyclooxygenase activity. Prostag Leukotr Ess 2000; 63: 371–375
Smith JA, Davis CL, Burgess GM . Prostaglandin E2-induced sensitization of bradykinin-evoked responses in rat dorsal root ganglion neurons is mediated by cAMP-dependent protein kinase A. Eur J Neurosci 2000; 12: 3250–3258
Hata Y, Clermont A, Yamauchi T, Pierce EA, Suzuma I, Kagokawa H et al. Retinal expression, regulation, and functional bioactivity of prostacyclin-stimulating factor. J Clin Invest 2000; 106: 541–550
Sato T, Sawada S, Tsuda Y, Komatsu S, Akamatsu N, Kono Y et al. The mechanism of thrombin-induced prostacyclin synthesis in human endothelial cells with reference to the gene transcription of prostacyclin-related enzymes and Ca2+ kinetics. J Pharmacol Toxicol Meth 1999; 41: 173–182
Nagao T, Illiano S, Vanhoutte PM . Heterogeneous distribution of endothelium-dependent relaxations resistant to NG-nitro-L-arginine in rats. Am J Physiol 1992; 263: H1090–H1094
Campbell WB, Gebremedhin D, Pratt PF, Harder DR . Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res 1996; 78: 415–423
Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Buss R . Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 1999; 401: 493–497
Fulton D, McGiff JC, Quilley J . Pharmacological evaluation of an epoxide as the putative hyperpolarizing factor mediating the nitric oxide-independent vasodilator effect of bradykinin in the rat heart. J Pharmacol Exp Ther 1998; 287: 497–503
Gebremedhin D, Harder DR, Pratt PF, Campbell WB . Bioassay of an endothelium-derived hyperpolarizing factor from bovine coronary arteries: role of a cytochrome P450 metabolite. J Vasc Res 1998; 35: 274–284
Hecker M, Bara AT, Bauersachs J, Busse R . Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J Physiol 1994; 481: 407–414
Rosolowsky M, Campbell WB . Role of PG12 and epoxyeicosatrienoic acids in relaxation of bovine coronary arteries to arachidonic acid. Am J Physiol 1993; 264: H327–H335
Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest 2000; 106: 1521–1530
Knot HJ, Zimmermann PA, Nelson MT . Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol 1996; 492: 419–430
Chaytor AT, Martin PE, Edwards DH, Griffith TM . Gap junctional communication underpins EDHF-type relaxations evoked by ACh in the rat hepatic artery. Am J Physiol Heart Circ Physiol 2001; 280: H2441–H2450
De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH, Vanhoutte PM . Endothelial dysfunction in diabetes. Br J Pharmacol 2000; 130: 963–974
Kiowski W, Luscher TF, Linder L, Buhler FR . Endothelin-1-induced vasoconstriction in humans. Reversal by calcium channel blockade but not by nitrovasodilators or endothelium-derived relaxing factor. Circulation 1991; 83: 469–475
Wang HG, Shibamoto T, Miyahara T . Endothelin-1 selectively contracts portal vein through both ETA and ETB receptors in isolated rabbit liver. Am J Physiol 1997; 273: G1036–G1043
Ito Y, Katori M, Majima M, Kakita A . Constriction of mouse hepatic venules and sinusoids by endothelins through ETB receptor subtype. Int J Microcirc Clin Exp 1996; 16: 250–258
Dehouck MP, Vigne P, Torpier G, Breittmayer JP, Cecchelli R, Frelin C . Endothelin-1 as a mediator of endothelial cell–pericyte interactions in bovine brain capillaries. J Cereb Blood Flow Metab 1997; 17: 464–469
Goto K, Kasuya Y, Matsuki N, Takuwa Y, Kurihara H, Ishikawa T et al. Endothelin activates the dihydropyridine-sensitive, voltage-dependent Ca2+ channel in vascular smooth muscle. Proc Natl Acad Sci U S A 1989; 86: 3915–3918
Resink TJ, Scott-Burden T, Buhler FR . Activation of phospholipase A2 by endothelin in cultured vascular smooth muscle cells. Biochem Biophys Res Commun 1989; 158: 279–286
Hirata Y, Yoshimi H, Takata S, Watanabe TX, Kumagai S, Nakajima K, Sakakibara S . Cellular mechanism of action by a novel vasoconstrictor endothelin in cultured rat vascular smooth muscle cells. Biochem Biophys Res Commun 1988; 154: 868–875
Chakrabarti S, Cukiernik M, Hileeto D, Evans T, Chen S . Role of vasoactive factors in the pathogenesis of early changes in diabetic retinopathy. Diabetes Metab Res Rev 2000; 16: 393–407
Abdel-Latif AA, Husain S, Yousufzai SY . Role of protein kinase C alpha and mitogen-activated activated protein kinases in endothelin-1-stimulation of cytosolic phospholipase A2 in iris sphincter smooth muscle. J Cardiovasc Pharmacol 2000; 36 (Suppl 5): S117–S119
Husain S, Abdel-Latif AA . Endothelin-1 activates p38 mitogen-activated protein kinase and cytosolic phospholipase A2 in cat iris sphincter smooth muscle cells. Biochem J 1999; 342: 87–96
Li P, Ferrario CM, Brosnihan KB . Losartan inhibits thromboxane A2-induced platelet aggregation and vascular constriction in spontaneously hypertensive rats. J Cardiovasc Pharmacol 1998; 32: 198–205
Minhas S, Eardley I, Joyce AD, Morrison JB . The effect of cyclic GMP on rabbit corporal smooth muscle tone and its modulation by cyclo-oxygenase products. Prostaglandins Leukot Essent Fatty Acids 2000; 62: 153–160
Katusic ZS, Vanhoutte PM . Superoxide anion is an endothelium-derived contracting factor. Am J Physiol 1989; 257: H33–H37
Bohlen HG, Lash JM . Topical hyperglycemia rapidly suppresses EDRF-mediated vasodilation of normal rat arterioles. Am J Physiol 1993; 265: H219–H225
Wen FQ, Watanabe K, Yoshida M . Eicosanoid profile in cultured human pulmonary artery smooth muscle cells treated with IL-1 beta and TNF alpha. Prostaglandins Leukot Essent Fatty Acids 1998; 59: 71–75
Kador PF, Takahashi Y, Wyman M, Ferris F 3rd . Diabetes-like proliferative retinal changes in galactose-fed dogs. Arch Ophthalmol 1995; 113: 352–354
Kerty E, Russell D, Bakke SJ, Nyberg-Hansen R, Rootwell K . Regional cerebral blood flow (rCBF) and cerebral vasoreactivity in patients with retinal ischaemic symptoms. J Neurol Neurosurg Psychiatry 1989; 52: 1345–1350
Naruse K, Nakamura J, Hamada Y, Nakayama M, Chaya S, Komori T et al. Aldose reductase inhibition prevents glucose-induced apoptosis in cultured bovine retinal microvascular pericytes. Exp Eye Res 2000; 71: 309–315
Iannello S, Cavaleri A, Camuto M, Belfiore F . In vitro inhibition of glucose phosphorylation by an aldose-reductase inhibitor (Tolrestat) in some non-insulin-sensitive rabbit tissues. J Diabetes Complications 1999; 13: 68–73
Segawa M, Hirata Y, Fujimori S, Okada K . The development of electroretinogram abnormalities and the possible role of polyol pathway activity in diabetic hyperglycemia and galactosemia. Metabolism 1988; 37: 454–460
Robison WG Jr, McBaleb ML, Feld LG, Michaelis OE, Laver N, Mercandetti M, Boninson WG . Degenerated intramural pericytes (‘ghost cells’) in the retinal capillaries of diabetic rats. Curr Eye Res 1991; 10: 339–350
Goldfarb S, Ziyadeh FN, Kern EF, Simmons DA . Effects of polyol-pathway inhibition and dietary myo-inositol on glomerular hemodynamic function in experimental diabetes mellitus in rats. Diabetes 1991; 40: 465–471
Narayanan S . Aldose reductase and its inhibition in the control of diabetic complications. Ann Clin Lab Sci 1993; 23: 148–158
Stevens MJ, Henry DN, Thomas TP, Killen PD, Greene DA . Aldose reductase gene expression and osmotic dysregulation in cultured human retinal pigment epithelial cells. Am J Physiol 1993; 265: E428–E438
Lim SS, Jung SH, Ji J, Shin KH, Keum SR . Synthesis of flavenoids and their effects on aldose reductase and and sorbitol accumulation in streptozotocin-induced diabetic rat tissues. J Pharm Pharmacol 2001; 53: 653–668
Sato S, Secchi EF, Lizak MJ, Fukase S, Ohta N, Murata M et al. Polyol formation and NADPH-dependent reductases in dog retinal capillary pericytes and endothelial cells. Invest Ophthalmol Vis Sci 1999; 40: 697–704
Bravi MC, Pietrangeli P, Laurenti O, Basili S, Cassone-Faldetta M, Ferri C, De Mattia G . Polyol pathway activation and glutathione redox status in non-insulin-dependent diabetic patients. Metabolism 1997; 46: 1194–1198
Kishi Y, Schmelzer JD, Yao JK, Zollman PJ, Nickander KK, Tritschler HJ, Low PA . Alpha-lipoic acid: effect on glucose uptake, sorbitol pathway, and energy metabolism in experimental diabetic neuropathy. Diabetes 1999; 48: 2045–2051
Dunn JA, McCance DR, Thorpe SR, Lyons TJ, Baynes JW . Age-dependent accumulation of N epsilon-(carboxymethyl)lysine and N epsilon-(carboxymethyl)hydroxylysine in human skin collagen. Biochemistry 1991; 30: 1205–1210
Khalifah RG, Baynes JW, Hudson BG . Amadorins: novel post-Amadori inhibitors of advanced glycation reactions. Biochem Biophys Res Commun 1999; 257: 251–258
Stitt AW . Advanced glycation: an important pathological event in diabetic and age related ocular disease. Br J Ophthalmol 2001; 85: 746–753
Gurler B, Vural H, Yilmaz N, Oguz H, Satici A, Aksoy N . The role of oxidative stress in diabetic retinopathy. Eye 2000; 14: 730–735
Agardh E, Hultberg B, Agardh C . Effects of inhibition of glycation and oxidative stress on the development of cataract and retinal vessel abnormalities in diabetic rats. Curr Eye Res 2000; 21: 543–549
Kern TS, Kowluru RA, Engerman RL . Abnormalities of retinal metabolism in diabetes or galactosemia: ATPases and glutathione. Invest Ophthalmol Vis Sci 1994; 35: 2962–2967
Roginsky V, Barsukova T . Superoxide dismutase inhibits lipid peroxidation in micelles. Chem Phys Lipids 2001; 111: 87–91
Sozmen EY, Sozmen B, Delen Y, Onat T . Catalase/superoxide dismutase (sod) and catalase/paraoxonase (pon) ratios may implicate poor glycemic control. Arch Med Res 2001; 32: 283–287
Mates JM, Perez-Gomez C, Nunez de Castro I . Antioxidant enzymes and human diseases. Clin Biochem 1999; 32: 595–603
Agardh CD, Agardh E, Hultberg B, Qian Y, Ostenson CG . The glutathione levels are reduced in Goto-Kakizaki rat retina, but are not influenced by aminoguanidine treatment. Curr Eye Res 1998; 17: 251–256
Kowluru RN, Keern TS, Engerman RL . Abnormalities of retinal metabolism in diabetes or experimental galactosemia. IV. Antioxidant defense system. Free Radic Biol Med 1997; 22: 587–592
Nourooz-Zadeh J, Pereira P . F(2) isoprostanes, potential specific markers of oxidative damage in human retina. Ophthalmic Res 2000; 32: 133–137
Bohlen HG, Lash JM . Topical hyperglycemia rapidly suppresses EDRG-mediated vasodilation of normal rat arterioles. Am J Physiol 1993; 265: H210–H225
Haefliger IO, Flammer J, Beny JL, Luscher TF . Endothelium-dependent vasoactive modulation in the ophthalmic circulation. Prog Retin Eye Res 2001; 20: 209–225
Moller P, Loft S, Lundby C, Olsen NV . Acute hypoxia and hypoxic exercise induce DNA strand breaks and oxidative DNA damage in humans. FASEB J 2001; 15: 1181–1186
Hinokio Y, Suzuki S, Hirai M, Chiba M, Hirai A, Toyota T . Oxidative DNA damage in diabetes mellitus: its association with diabetic complications. Diabetologia 1999; 42: 995–998
Li W, Yanoff M, Jian B, He Z . Altered mRNA levels of antioxidant enzymes in pre-apoptotic pericytes from human diabetic retinas. Cell Mol Biol 1999; 45: 59–66
Podesta F, Romeo G, Liu WH, Krajewski S, Reed JC, Gerhardinger C, Lorenzi M . Bax is increased in the retina of diabetic subjects and is associated with pericyte apoptosis in vivo and in vitro. Am J Pathol 2000; 156: 1025–1032
Li W, Yanoff M, Liu X, Ye X . Retinal capillary pericyte apoptosis in early human diabetic retinopathy. Chin Med J (Engl) 1997; 110: 659–663
Kern TS, Engerman RL . Capillary lesions develop in retina rather than cerebral cortex in diabetes and experimental galactosemia. Arch Ophthalmol 1996; 114: 306–310
Kern TS, Engerman RL . Vascular lesions in diabetes are distributed non-uniformly within the retina. Exp Eye Res 1995; 60: 545–549
Stitt AW, Gardiner TA, Archer DB . Histological and ultrastructural investigation of retinal microaneurysm development in diabetic patients. Br J Ophthalmol 1995; 79: 362–367
Nishikawa T, Giardino I, Edelstein D, Brownlee M . Changes in diabetic retinal matrix protein mRNA levels in common transgenic mouse strain. Curr Eye Res 2000; 21: 581–587
Bek T, Ledet T . Glycoprotein deposition in vascular walls of diabetic retinopathy. A histopathological and immunohistochemical study. Acta Ophthalmol Scand 1996; 74: 385–390
Jerdan JA, Michels RG, Glaser BM . Diabetic preretinal membranes. An immunohisochemical study. Arch Opthalmol 1986; 104: 286–290
Casaroli Marano RP, Preissner KT, Vilaro S . Fibronectin, laminin, vitronectin and their receptors at newly-formed capillaries in proliferative diabetic retinopathy. Exp Eye Res 1995; 60: 5–17
Roy S, Lorenzi M . Early biosynthetic changes in the diabetic-like retinopathy of galactose-fed rats. Diabetologia 1996; 39: 735–738
Hosoda Y, Okada M, Matsumura M, Ogino N, Honda Y, Nagai Y . Epiretinal membrane of proliferative diabetic retinopathy: an immunohistochemical study. Ophthalmic Res 1993; 25: 289–294
Abrass CK, Peterson CV, Raugi GJ . Phenotypic expression of collagen types in mesangial matrix of diabetic and nondiabetic rats. Diabetes 1988; 37: 1695–1702
Spirin KS, Saghizadeh M, Lewin SL, Zardi L, Kenney MC, Ljubimov AV . Basement membrane and growth factor gene expression in normal and diabetic human retinas. Curr Eye Res 1999; 18: 490–499
Jian B, Jones PL, Li Q, Mohler ER 3rd, Schoen FJ, Levy RJ . Matrix metalloproteinase-2 is associated with tenascin-c in calcific aortic stenosis. Am J Pathol 2001; 159: 321–327
Boeri D, Maiello M, Lorenzi M . Increased prevalence of microthromboses in retinal capillaries of diabetic individuals. Diabetes 2000; 50: 1432–1439
Lee IK, Koya D, Ishi H, Kanoh H, King GL . d-Alpha-tocopherol prevents the hyperglycemia induced activation of diacylglycerol (DAG)-protein kinase C (PKC) pathway in vascular smooth muscle cells by an increase of DAG kinase activity. Diabetes Res Clin Pract 1999; 45: 183–190
Takahashi T, Hato F, Yamane T, Fukumasu H, Suzuki K, Ogita S et al. Activation of human neutrophils by cytokine-activated endothelial cells. Circ Res 2001; 88: 422–429
de la Cruz JP, Moreno A, Ruiz-Ruiz MI, Garcia-Campos J, Sanchez de la Cuesta F . Effect of WEB 2086-BS, an antagonist of platelet-activating factor receptors, on retinal vascularity in diabetic rats. Eur J Pharmacol 1998; 360: 37–42
Bussolino F, Gremo F, Tetta C, Pescarmona GP, Camussi G . Production of platelet-activating factor by chick retina. J Biol Chem 1986; 261: 16502–16508
Thierry A, Doly M, Braquet P, Cluzel J, Meyniel G . Presence of specific platelet-activating factor binding sites in the rat retina. Eur J Pharmacol 1989; 163: 97–101
Omoto S, Nomura S, Shouzu A, Hayakawa T, Shimizu H, Miyake Y et al. Significance of platelet-derived microparticles and activated platelets in diabetic nephropathy. Nephron 1999; 81: 271–277
Nomura S, Nakamura T, Cone J, Tandon NN, Kambayashi J . Cytometric analysis of high shear-induced platelet microparticles and effect of cytokines on microparticle generation. Cytometry 2000; 40: 173–181
Li M, Goto S, Sakai H, Kim JY, Ichikawa N, Yoshida M et al. Enhanced shear-induced von Willebrand factor binding to platelets in acute myocardial infarction. Thromb Res 2000; 100: 251–261
De la Cruz JP, Maximo MA, Blanco E, Moreno A, Sanchez de la Cuesta F . Effect of erythrocytes and prostacyclin production in the effect of fructose and sorbitol on platelet activation in human whole blood in vitro. Thromb Res 1997; 86: 515–524
Phillips AO, Morrisey K, Steadman R, Williams JD . Decreased degradation of collagen and fibronectin following exposure of proximal cells to glucose. Exp Nephrol 1999; 7: 449–462
De La Cruz JP, Moreno A, Ruiz-Ruiz MI, Garcia Campos J, Sanchez de la Cuesta F . Effect of camonagrel, a selective thromboxane synthase inhibitor, on retinal vascularization in experimental diabetes. Eur J Pharmacol 1998; 350: 81–85
Haimovich B, Lipfert L, Brugge JS, Shattil SJ . Tyrosine phosphorylation and cytoskeletal reorganization in platelets are triggered by interaction of integrin receptors with their immobilized ligands. J Biol Chem 1993; 268: 15868–15877
Miyamoto K, Khosrof S, Bursell SE, Rohan R, Murata T, Clermont AC et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A 1999; 96: 10836–10841
Hayashi M, Imai Y, Oh-ishi S . Phorbol ester stimulates PAF synthesis via the activation of protein kinase C in rat leukocytes. Lipids 1991; 26: 1054–1059
Whatley RE, Nelson P, Zimmerman GA, Stevens DL, Parker CJ, McIntyre TM, Prescott SM . The regulation of platelet-activating factor production in endothelial cells. The role of calcium and protein kinase C. J Biol Chem 1989; 264: 6325–6333
Okayama N, Coe L, Itoh M, Alexander JS . Exogenous nitric oxide increases neutrophil adhesion to cultured human endothelial monolayers through a protein kinase G dependent mechanism. Inflammation 1999; 23: 37–50
Triggiani M, Oriente A, Golino P, Gentile M, Battaglia C, Brevetti G, Marone G . Inhibition of platelet-activating factor synthesis in human neutrophils and platelets by propionyl-L-carnitine. Biochem Pharmacol 1999; 58: 1341–1348
Komatsu H, Amano M, Yamaguchi S, Sugahara K . Inhibition of activation of human peripheral blood eosinophils by Y-24180, an antagonist to platelet-activating factor receptor. Life Sci 1999; 65: PL171–PL176
Kubes P, Grisham MB, Barrowman JA, Gaginella T, Granger DN . Leukocyte-induced vascular protein leakage in cat mesentery. Am J Physiol 1991; 261: H1872–H1879
Zimmerman GA, McIntyre TM, Prescott SM . Thrombin stimulates the adherence of neutrophils to human endothelial cell in vitro. Immunol Today 1992; 13: 93–99
Granger DN, Kubes P . The microcirculation and inflammation: modulation of leukocyte-enothelial cell adhesion. J Leukoc Biol 1994; 55: 662–675
Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP . Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 2001; 158: 147–152
Lane TA, Lamkin GE, Wancewicz E . Modulation of endothelial cell expression of intercellular adhesion molecule 1 by protein kinase C activation. Biochem Biophys Res Commun 1989; 161: 945–952
Wertheimer SJ, Myers CL, Wallace RW, Parks TP . Intercellular adhesion molecule-1 gene expression in human endothelial cells. Differential regulation by tumor necrosis factor-alpha and phorbol myristate acetate. J Biol Chem 1992; 267: 12030–12035
Arndt H, Russell JB, Kurose I, Kubes P, Granger DN . Mediators of leukocyte adhesion in rat mesenteric venules elicited by inhibition of nitric oxide synthesis. Gastroenterology 1993; 105: 675–680
Kubes P, Suzuki M, Granger DN . Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA 1991; 88: 4651–4655
Worthen GS, Schwab B 3rd, Elson EL, Downey GP . Mechanics of stimulated neutrophils: cell stiffening induces retention in capillaries. Science 1989; 245: 183–186
Kinukawa Y, Shimura M, Tamai M . Quantifying leukocyte dynamics and plugging in retinal microcirculation of streptozotosin-induced diabetic rats. Curr Eye Res 1999; 18: 49–55
Meisel SR, Shapiro H, Radnay J, Neuman Y, Khaskia AR, Gruener N et al. Increased expression of neutrophil and monocyte adhesion molecules LFA-1 and Mac-1 and their ligand ICAM-1 and VLA-4 throughout the acute phase of myocardial infarction: possible implications for leukocyte aggregation and microvascular plugging. J Am Coll Cardiol 1998; 31: 120–125
Schroder S, Palinski W, Schmid-Schonbein GW . Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol 1991; 139: 81–100
Wallow IH, Danis RP, Bindley C, Neider M . Cystoid macular degeneration in experimental branch retinal vein occlusion. Ophthalmology 1988; 95: 1371–1379
Bellhorn RW, Burns MS, Benjamin JV . Retinal vessel abnormalities of phototoxic retinopathy in rats. Invest Ophthalmol Vis Sci 1980; 19: 584–595
Lu M, Kuroki M, Amano S, Tolentino M, Keough K, Kim I et al. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest 1998; 101: 1219–1224
Gries FA . Alternative therapeutic principles in the prevention of microvascular and neuropathic complications. Diabetes Res Clin Pract 1995; 28 (Suppl): S201–S207
Michiels C, Arnould T, Remacle J . Endothelial cell responses to hypoxia: initiation of a cascade of cellular interactions. Biochim Biophys Acta 2000; 1497: 1–10
Tailor A, Granger DN . Role of adhesion molecules in vascular regulation and damage. Curr Hypertens Rep 2000; 2: 78–83
Sivalingam A, Kenney J, Brown GC, Benson WE, Donoso L . Basic fibroblast growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol 1990; 108: 869–872
Michaelson I . The mode of development of the vascular system of the retina with observations on its significance for certain retinal diseases. Trans Ophthalmol Soc UK 1948; 68: 137–180
Ashton N . Retinal vascularization in health and disease. Am J Ophthalmol 1957; 44: 7–17
Lahrmann C, Bek T . Foveal haemorrhages in diabetic retinopathy. Clinical characteristics and visual outcome. Acta Ophthalmol Scand 2000; 78: 169–172
Garner A . Histopathology of diabetic retinopathy in man. Eye 1993; 7: 250–253
Stefansson E . Laser treatment in diabetes-related eye complications. Nord Med 1992; 107: 309–310
Scott IU, Flynn HW Jr, Hughes JR . Echographic evaluation of a patient with diabetes and dense vitreous hemorrhage: an avulsed retinal vessel may mimic a tractional retinal detachment. Am J Ophthalmol 2001; 131: 515–516
Zhu H, Bunn HF . Oxygen sensing and signaling: impact on the regulation of physiologically important genes. Respir Physiol 1999; 115: 239–247
Zhu H, Riggs AF . Yeast flavohemoglobin is an ancient protein related to globins and a reductase family. Proc Natl Acad Sci U S A 1992; 89: 5015–5019
Caro J . Hypoxia regulation of gene transcription. High Alt Med Biol 2001; 2: 145–154
Zhong H, Hanrahan C, van der Poel H, Simons JW . Hypoxia-inducible factor 1 alpha and 1 beta proteins share common signaling pathways in human prostate cancer cells. Biochem Biophys Res Commun 2001; 284: 352–356
Mazure NM, Chen EY, Laderoute KR, Giaccia AJ . Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood 1997; 90: 3322–3331
Ozaki H, Yu AY, Della N, Ozaki K, Luna JD, Yamada H et al. Hypoxia inducible factor-1 alpha is increased in ischemic retina: temporal and spatial correlation with VEGF expression. Invest Ophthalmol Vis Sci 1999; 40: 182–189
Ferrara N, Henzel WJ . Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun 1989; 161: 851–858
Levy AP, Levy NS, Wegner S, Goldberg MA . Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem 1995; 270: 13333–13340
Williams B . Vascular permeability/vascular endothelial growth factors: a potential role in the pathogenesis and treatment of vascular diseases. Vasc Med 1996; 1: 251–258
Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 1994; 331: 1480–1487
Ferrara N . Role of vascular endothelial growth factor in the regulation of angiogenesis. Kidney Int 1999; 56: 794–814
Mesri M, Morales-Ruiz M, Ackermann EJ, Bennett CF, Pober JS, Sessa WC, Altieri DC . Suppression of vascular endothelial growth factor-mediated endothelial cell protection by surviving targeting. Am J Pathol 2001; 158: 1757–1765
de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT . The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992; 255: 989–991
Terman BI, Dougher-Vermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D, Bohlen P . Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun 1992; 187: 1579–1586
Shibuya M, Yamaguchi S, Yamane A, Ikeda T, Tojo A, Matsushime H, Sato M . Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene 1990; 5: 519–524
Terman BI, Carrion ME, Kovacs E, Rasmussen BA, Eddy RL, Shows TB . Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene 1991; 6: 1677–1683
Matthews W, Jordan CT, Gavin M, Jenkins NA, Copeland NG, Lemischka IR . A receptor tyrosine kinase cDNA isolated from a population of enriched primitive hematopoietic cells and exhibiting close genetic linkage to c-kit. Proc Natl Acad Sci U S A 1991; 88: 9026–9030
Quinn TP, Peters KG, De Vries C, Ferrara N, Williams LT . Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium. Proc Natl Acad Sci U S A 1993; 90: 7533–7537
Millauer B, Wizigmann-Voos S, Schnurch H, Martinez R, Moller NP, Risau W, Ullrich A . High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 1993; 72: 835–846
Takahashi T, Yamaguchi S, Chida K, Shibuya M . A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J 2001; 20: 2768–2778
Takahashi T, Shibuya M . The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-gamma pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene 1997; 14: 2079–2089
Qi JH, Claesson-Welsh L . VEGF-induced activation of phosphoinositide 3-kinase is dependent on focal adhesion kinase. Exp Cell Res 2001; 263: 173–182
Suzuma K, Naruse K, Suzuma I, Takahara N, Ueki K, Aiello LP, King GL . Vascular endothelial growth factor induces expression of connective tissue growth factor via KDR, Flt1, and phosphatidylinositol 3-kinase-akt-dependent pathways in retinal vascular cells. J Biol Chem 2000; 275: 40725–40731
Enaida H, Kabuyama Y, Oshima Y, Sakamoto T, Kato K, Kochi H, Homma Y . VEGF-dependent signaling in retinal microvascular endothelial cells. Fukushima J Med Sci 1999; 45: 77–91
Igal RA, Caviglia JM, de Gomez Dumm IN, Coleman RA . Diacylglycerol generated in CHO cell plasma membrane by phospholipase C is used for triacylglycerol synthesis. J Lipid Res 2001; 42: 88–95
Kim MJ, Kim E, Ryu SH, Suh PG . The mechanism of phospholipase C-gamma1 regulation. Exp Mol Med 2000; 32: 101–109
Boulton M, Foreman D, Williams G, McLeod D . VEGF localisation in diabetic retinopathy. Br J Ophthalmol 1998; 82: 561–568
Adamis AP, Miller JW, Bernal MT, D’Amico DJ, Folkman J, Yeo TK, Yeo KT . Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol 1994; 118: 445–450
Malecaze F, Clamens S, Simorre-Pinatel V, Mathis A, Chollet P, Favard C et al. Detection of vascular endothelial growth factor messenger RNA and vascular endothelial growth factor-like activity in proliferative diabetic retinopathy. Arch Ophthalmol 1994; 112: 1476–1482
Segawa Y, Shirao Y, Yamagishi S, Higashide T, Kobayashi M, Katsuno K et al. Upregulation of retinal vascular endothelial growth factor mRNAs in spontaneously diabetic rats without ophthalmoscopic retinopathy. A possible participation of advanced glycation end products in the development of the early phase of diabetic retinopathy. Ophthalmic Res 1998; 30: 333–339
Simpson DA, Murphy GM, Bhaduri T, Gardiner TA, Archer DB, Stitt AW . Expression of the VEGF gene family during retinal vaso-obliteration and hypoxia. Biochem Biophys Res Commun 1999; 262: 333–340
Grishko V, Solomon M, Breit JF, Killilea DW, Ledoux SP, Wilson GL, Gillespie MN . Hypoxia promotes oxidative base modifications in the pulmonary artery endothelial cell VEGF gene. FASEB J 2001; 15: 1267–1269
Smith G, McLeod D, Foreman D, Boulton M . Immunolocalisation of the VEGF receptors FLT-1, KDR, and FLT-4 in diabetic retinopathy. Br J Ophthalmol 1999; 83: 486–494
Gilbert RE, Vranes D, Berka JL, Kelly DJ, Cox A, Wu LL et al. Vascular endothelial growth factor and its receptors in control and diabetic rat eyes. Lab Invest 1998; 78: 1017–1027
Amin RH, Frank RN, Kennedy A, Eliott D, Puklin JE, Abrams GW . Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy. Invest Ophthalmol Vis Sci 1997; 38: 36–47
Sone H, Kawakami Y, Okuda Y, Sekine Y, Honmura S, Matsuo K et al. Ocular vascular endothelial growth factor levels in diabetic rats are elevated before observable retinal proliferative changes. Diabetologia 1997; 40: 726–730
Clermont AC, Aiello LP, Mori F, Aiello LM, Bursell SE . Vascular endothelial growth factor and severity of nonproliferative diabetic retinopathy mediate retinal hemodynamics in vivo: a potential role for vascular endothelial growth factor in the progression of nonproliferative diabetic retinopathy. Am J Ophthalmol 1997; 124: 433–446
Tolentino MJ, Miller JW, Gragoudas ES, Jakobiec FA, Flynn E, Chatzistefanou K et al. Intravitreous injections of vascular endothelial growth factor produce retinal ischemia and microangiopathy in an adult primate. Ophthalmology 1996; 103: 1820–1828
Kevil CG, Payne DK, Mire E, Alexander JS . Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem 1998; 273: 15099–15103
Esser S, Lampugnani MG, Corada M, Dejana E, Risau W . Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 1998; 111: 1853–1865
Yoshida A, Feke GT, Morales-Stoppello J, Collas GD, Goger DG, McMeel JW . Retinal blood flow alterations during progression of diabetic retinopathy. Arch Ophthalmol 1983; 101: 225–227
Robinson GS, Pierce EA, Rook SL, Foley E, Webb R, Smith LE . Oligodeoxynucleotides inhibit retinal neovascularization in a murine model of proliferative retinopathy. Proc Natl Acad Sci U S A 1996; 93: 4851–4856
Adamis AP, Shima DT, Tolentino MJ, Gragoudas ES, Ferrara N, Folkman J et al. Inhibition of vascular endothelial growth factor prevents retinal ischemia-associated iris neovascularization in a nonhuman primate. Arch Ophthalmol 1996; 114: 66–71
Aiello LP, Pierce EA, Foley ED, Takagi H, Chen H, Riddle L et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci U S A 1995; 92: 10457–10461
Poulsen JE . Recovery from retinopathy in a case of diabetes with Simmond’s disease. Diabetes 1953; 2: 7–12
Smith LE, Kopchick JJ, Chen W, Knapp J, Kinose F, Daley D et al. Essential role of growth hormone in ischemia-induced retinal neovascularization. Science 1997; 276: 1706–1709
Hyer SL, Sharp PS, Brooks RA, Burrin JM, Kohner EM . A two-year follow-up study of serum insulinlike growth factor-I in diabetics with retinopathy. Metabolism 1989; 38: 586–589
Hyer SL, Sharp PS, Brooks RA, Burrin JM, Kohner EM . Serum IGF-1 concentration in diabetic retinopathy. Diabet Med 1988; 5: 356–360
Grant MB, Mames RN, Fitzgerald C, Ellis EA, Aboufriekha M, Guy J . Insulin-like growth factor I acts as an angiogenic agent in rabbit cornea and retina: comparative studies with basic fibroblast growth factor. Diabetologia 1993; 36: 282–291
Lee HC, Lee KW, Chung CH, Chung YS, Lee EJ, Lim SK et al. IGF-I of serum and vitreous fluid in patients with diabetic proliferative retinopathy. Diabetes Res Clin Pract 1994; 24: 85–88
Nakao-Hayashi J, Ito H, Kanayasu T, Morita I, Murota S . Stimulatory effects of insulin and insulin-like growth factor I on migration and tube formation by vascular endothelial cells. Atherosclerosis 1992; 92: 141–149
King GL, Goodman AD, Buzney S, Moses A, Kahn CR . Receptors and growth promoting effects of insulin and insulin-like growth factors on cells from bovine retinal capillaries and aorta. J Clin Invest 1985; 75: 1028–1036
Nicosia RF, Nicosia SV, Smith M . Vascular endothelial growth factor, platelet-derived growth factor, and insulin-like growth factor-1 promote rat aortic angiogenesis in vitro. Am J Pathol 1994; 145: 1023–1029
Grant MB, Mames RN, Fitzgerald C, Ellis EA, Aboufriekha M, Guy J . Insulin-like growth factor I acts as an angiogenic agent in rabbit cornea and retina: comparative studies with basic fibroblast growth factor. Diabetologia 1993; 36: 282–291
Boney CM, Sekimoto H, Gruppuso PA, Frackelton AR Jr . Src family tyrosine kinases participate in insulin-like growth factor in mitogenic signaling in 3t3-11 cells. Cell Growth Differ 2001; 12: 379–386
Shoba LN, Newman M, Liu W, Lowe WL Jr . LY 294002, an inhibitor of phosphatidylinositol 3-kinase, inhibits GH-mediated expression of the IGF-I gene in rat hepatocytes. Endocrinology 2001; 142: 3980–3986
Yamada M, Tanabe K, Wada K, Shimoke K, Ishikawa Y, Ikeuchi T et al. Differences in survival-promoting effects and intracellular signaling properties of BDNF and IGF-1 in cultured cerebral cortical neurons. J Neurochem 2001; 78: 940–951
Arsenijevic Y, Weiss S, Schneider B, Aebischer P . Insulin-like growth factor-I is necessary for neural stem cell proliferation and demonstrates distinct actions of epidermal growth factor and fibroblast growth factor-2. J Neurosci 2001; 21: 7194–7202
Burren CP, Berka JL, Edmondson SR, Werther GA, Batch JA . Localization of mRNAs for insulin-like growth factor-I (IGF-I), IGF-I receptor, and IGF binding proteins in rat eye. Invest Ophthalmol Vis Sci 1996; 37: 1459–1468
Tollefsen SE, Heath-Monnig E, Cascieri MA, Bayne ML, Daughaday WH . Endogenous insulin-like growth factor (IGF) binding proteins cause IGF-1 resistance in cultured fibroblasts from a patient with short stature. J Clin Invest 1991; 87: 1241–1250
Meyer-Schwickerath R, Pfeiffer A, Blum WF, Freyberger H, Klein M, Losche C et al. Vitreous levels of the insulin-like growth factors I and II, and the insulin-like growth factor binding proteins 2 and 3, increase in neovascular eye disease. Studies in nondiabetic and diabetic subjects. J Clin Invest 1993; 92: 2620–2625
Boulton M, Gregor Z, McLeod D, Charteris D, Jarvis-Evans J, Moriarty P et al. Intravitreal growth factors in proliferative diabetic retinopathy: correlation with neovascular activity and glycaemic management. Br J Ophthalmol 1997; 81: 228–233
Burgos R, Mateo C, Canton A, Hernandez C, Mesa J, Simo R . Vitreous levels of IGF-I, IGF binding protein 1, and IGF binding protein 3 in proliferative diabetic retinopathy: a case-control study. Diabetes Care 2000; 23: 80–83
Spoerri PE, Ellis EA, Tarnuzzer RW, Grant MB . Insulin-like growth factor: receptor and binding proteins in human retinal endothelial cell cultures of diabetic and non-diabetic origin. Growth Horm IGF Res 1998; 8: 125–132
Tucci M, Nygard K, Tanswell BV, Farber HW, Hill DJ, Han VK . Modulation of insulin-like growth factor (IGF) and IGF binding protein biosynthesis by hypoxia in cultured vascular endothelial cells. J Endocrinol 1998; 157: 13–24
Tazuke SI, Mazure NM, Sugawara J, Carland G, Faessen GH, Suen LF et al. Hypoxia stimulates insulin-like growth factor binding protein 1 (IGFBP-1) gene expression in HepG2 cells: a possible model for IGFBP-1 expression in fetal hypoxia. Proc Natl Acad Sci USA 1998; 95: 10188–10193
Moriarty P, Boulton M, Dickson A, McLeod D . Production of IGF-I and IGF binding proteins by retinal cells in vitro. Br J Ophthalmol 1994; 78: 638–642
Presta M, Maier JA, Rusnati M, Ragnotti G . Basic fibroblast growth factor is released from endothelial extracellular matrix in a biologically active form. J Cell Physiol 1989; 140: 68–74
Globus RK, Plouet J, Gospodarowicz D . Cultured bovine bone cells synthesize basic fibroblast growth factor and store it in their extracellular matrix. Endocrinology 1989; 124: 1539–1547
Bashkin P, Doctrow S, Klagsbrun M, Svahn CM, Folkman J, Vlodavsky I . Basic fibroblast growth factor binds to subendothelial extracellular matrix and is released by heparitinase and heparin-like molecules. Biochemistry 1989; 28: 1737–1743
Stavri GT, Zachary IC, Baskerville PA, Martin JF, Erusalimsky JD . Basic fibroblast growth factor upregulates the expression of vascular endothelial growth factor in vascular smooth muscle cells. Synergistic interaction with hypoxia. Circulation 1995; 92: 11–4
Ozaki H, Okamoto N, Ortega S, Chang M, Ozaki K, Sadda S et al. Basic fibroblast growth factor is neither necessary nor sufficient for the development of retinal neovascularization. Am J Pathol 1998; 153: 757–765
Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES et al. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol 1998; 141: 1659–1673
Williams LT . Signal transduction by the platelet-derived growth factor receptor. Science 1989; 243: 1564–1570
Claesson-Welsh L . Platelet-derived growth factor receptor signals. J Biol Chem 1994; 269: 32023–32026
Kourembanas S, Hannan RL, Faller DV . Oxygen tension regulates the expression of the platelet-derived growth factor-B chain gene in human endothelial cells. J Clin Invest 1990; 86: 670–674
Kuwabara K, Ogawa S, Matsumoto M, Koga S, Clauss M, Pinsky DJ et al. Hypoxia-mediated induction of acidic/basic fibroblast growth factor and platelet-derived growth factor in mononuclear phagocytes stimulates growth of hypoxic endothelial cells. Proc Natl Acad Sci USA 1995; 92: 4606–4610
Freyberger H, Brocker M, Yakut H, Hammer J, Effert R, Schifferdecker E et al. Increased levels of platelet-derived growth factor in vitreous fluid of patients with proliferative diabetic retinopathy. Exp Clin Endocrinol Diabetes 2000; 108: 106–109
Koyama N, Watanabe S, Tezuka M, Morisaki N, Saito Y, Yoshida S . Migratory and proliferative effect of platelet-derived growth factor in rabbit retinal endothelial cells: evidence of an autocrine pathway of platelet-derived growth factor. J Cell Physiol 1994; 158: 1–6
Smits A, Hermansson M, Nister M, Karnushina I, Heldin CH, Westermark B, Funa K . Rat brain capillary endothelial cells express functional PDGF B-type receptors. Growth Factors 1989; 2: 1–8
Battegay EJ, Rupp J, Iruela-Arispe L, Sage EH, Pech M . PDGF-BB modulates endothelial proliferation and angiogenesis in vitro via PDGF beta-receptors. J Cell Biol 1994; 125: 917–928
Forsberg K, Valyi-Nagy I, Heldin CH, Herlyn M, Westermark B . Platelet-derived growth factor (PDGF) in oncogenesis: development of a vascular connective tissue stroma in xenotransplanted human melanoma producing PDGF-BB. Proc Natl Acad Sci USA 1993; 90: 393–397
Laterra J, Nam M, Rosen E, Rao JS, Lamszus K, Goldberg ID, Johnston P . Scatter factor/hepatocyte growth factor gene transfer enhances glioma growth and angiogenesis in vivo. Lab Invest 1997; 76: 565–577
Shinozuka H, Kubo Y, Katyal SL, Coni P, Ledda-Columbano GM, Columbano A, Nakamura T . Roles of growth factors and of tumor necrosis factor-alpha on liver cell proliferation induced in rats by lead nitrate. Lab Invest 1994; 71: 35–41
Pons E, Uphoff CC, Drexler HG . Expression of hepatocyte growth factor and its receptor c-met in human leukemia-lymphoma cell lines. Leuk Res 1998; 22: 797–804
He PM, He S, Garner JA, Ryan SJ, Hinton DR . Retinal pigment epithelial cells secrete and respond to hepatocyte growth factor. Biochem Biophys Res Commun 1998; 249: 253–257
Cai W, Rook SL, Jiang ZY, Takahara N, Aiello LP . Mechanisms of hepatocyte growth factor-induced retinal endothelial cell migration and growth. Invest Ophthalmol Vis Sci 2000; 41: 1885–1893
Grant DS, Kleinman HK, Goldberg ID, Bhargava MM, Nickoloff BJ, Kinsella JL et al. Scatter factor induces blood vessel formation in vivo. Proc Natl Acad Sci USA 1993; 90: 1937–1941
Lamszus K, Schmidt NO, Jin L, Laterra J, Zagzag D, Way D et al. Scatter factor promotes motility of human glioma and neuromicrovascular endothelial cells. Int J Cancer 1998; 75: 19–28
Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA . Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991; 251: 802–804
Shiota G, Kawasaki H, Nakamura T, Schmidt EV . Inhibitory effect of hepatocyte growth factor against FaO hepatocellular carcinoma cells may be associated with changes of intracellular signalling pathways mediated by protein kinase C. Res Commun Mol Pathol Pharmacol 1994; 85: 271–278
Graziani A, Gramaglia D, Cantley LC, Comoglio PM . The tyrosine-phosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. J Biol Chem 1991; 266: 22087–22090
Derman MP, Cunha MJ, Barros EJ, Nigam SK, Cantley LG . HGF-mediated chemotaxis and tubulogenesis require activation of the phosphatidylinositol 3-kinase. Am J Physiol 1995; 268: F1211–F1217
Morishita R, Nakamura S, Nakamura Y, Aoki M, Moriguchi A, Kida I et al. Potential role of an endothelium-specific growth factor, hepatocyte growth factor, on endothelial damage in diabetes. Diabetes 1997; 46: 138–142
Van Belle E, Witzenbichler B, Chen D, Silver M, Chang L, Schwall R, Isner JM . Potentiated angiogenic effect of scatter factor/hepatocyte growth factor via induction of vascular endothelial growth factor: the case for paracrine amplification of angiogenesis. Circulation 1998; 97: 381–390
Wojta J, Kaun C, Breuss JM, Koshelnick Y, Beckmann R, Hattey E et al. Hepatocyte growth factor increases expression of vascular endothelial growth factor and plasminogen activator inhibitor-1 in human keratinocytes and the vascular endothelial growth factor receptor flk-1 in human endothelial cells. Lab Invest 1999; 79: 427–438
Gille J, Khalik M, Konig V, Kaufmann R . Hepatocyte growth factor/scatter factor (HGF/SF) induces vascular permeability factor (VPF/VEGF) expression by cultured keratinocytes. J Invest Dermatol 1998; 111: 1160–1165
Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG . Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci USA 1991; 88: 9267–9271
Terman B, Khandke L, Dougher-Vermazan M, Maglione D, Lassam NJ, Gospodarowicz D et al. VEGF receptor subtypes KDR and FLT1 show different sensitivities to heparin and placenta growth factor. Growth Factors 1994; 11: 187–195
Landgren E, Schiller P, Cao Y, Claesson-Welsh L . Placenta growth factor stimulates MAP kinase and mitogenicity but not phospholipase C-gamma and migration of endothelial cells expressing Flt 1. Oncogene 1998; 16: 359–367
Seetharam L, Gotoh N, Maru Y, Neufeld G, Yamaguchi S, Shibuya M . A unique signal transduction from FLT tyrosine kinase, a receptor for vascular endothelial growth factor VEGF. Oncogene 1995; 10: 135–147
Ziche M, Maglione D, Ribatti D, Morbidelli L, Lago CT, Battisti M et al. Placenta growth factor-1 is chemotactic, mitogenic, and angiogenic. Lab Invest 1997; 76: 517–531
Cao Y, Chen H, Zhou L, Chiang MK, Anand-Apte B, Weatherbee JA et al. Heterodimers of placenta growth factor/vascular endothelial growth factor. Endothelial activity, tumor cell expression, and high affinity binding to Flk-1/KDR. J Biol Chem 1996; 271: 3154–3162
Yamashita H, Eguchi S, Watanabe K, Takeuchi S, Yamashita T, Miura M . Expression of placenta growth factor (PIGF) in ischaemic retinal diseases. Eye 1999; 13: 372–374
Khaliq A, Foreman D, Ahmed A, Weich H, Gregor Z, McLeod D, Boulton M . Increased expression of placenta growth factor in proliferative diabetic retinopathy. Lab Invest 1998; 78: 109–116
Spirin KS, Saghizadeh M, Lewin SL, Zardi L, Kenney MC, Ljubimov AV . Basement membrane and growth factor gene expression in normal and diabetic human retinas. Curr Eye Res 1999; 18: 490–499
Simpson DA, Murphy GM, Bhaduri T, Gardiner TA, Archer DB, Stitt AW . Expression of the VEGF gene family during retinal vaso-obliteration and hypoxia. Biochem Biophys Res Commun 1999; 262: 333–340
Yonekura H, Sakurai S, Liu X, Migita H, Wang H, Yamagishi S et al. Placenta growth factor and vascular endothelial growth factor B and C expression in microvascular endothelial cells and pericytes. Implication in autocrine and paracrine regulation of angiogenesis. J Biol Chem 1999; 274: 35172–35178
Tallquist MD, Soriano P, Klinghoffer RA . Growth factor signaling pathways in vascular development. Oncogene 1999; 18: 7917–7932
Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V et al. Isolation of angiopoietin-1, a ligand for the Tie2 receptor, by secretion-trap expression cloning. Cell 1996; 87: 1161–1169
Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997; 277: 55–60
Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S et al. Requisite role of angiopoietin-1, a ligand for the Tie2 receptor, during embryonic angiogenesis. Cell 1997; 87: 1171–1180
Hayes AJ, Huang WQ, Mallah J, Yang D, Lippman ME, Li LY . Angiopoietin-1 and its receptor Tie-2 participate in the regulation of capillary-like tubule formation and survival of endothelial cells. Microvasc Res 1999; 58: 224–237
Koblizek TI, Weiss C, Yancopoulos GD, Deutsch U, Risau W . Angiopoietin-1 induces sprouting angiogenesis in vitro. Curr Biol 1998; 8: 529–532
Kukk E, Wartiovaara U, Gunji Y, Kaukonen J, Buhring HJ, Rappold I et al. Analysis of Tie receptor tyrosine kinase in haemopoietic progenitor and leukaemia cells. Br J Haematol 1997; 98: 195–203
Stratmann A, Risau W, Plate KH . Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am J Pathol 1998; 153: 1459–1466
Hackett SF, Ozaki H, Strauss RW, Wahlin K, Suri C, Maisonpierre P et al. Angiopoietin 2 expression in the retina: upregulation during physiologic and pathologic neovascularization. J Cell Physiol 2000; 184: 275–284
Oh H, Takagi H, Suzuma K, Otani A, Matsumura M, Honda Y . Hypoxia and vascular endothelial growth factor selectively up-regulate angiopoietin-2 in bovine microvascular endothelial cells. J Biol Chem 1999; 274: 15732–15739
Tombran-Tink J, Chader GG, Johnson LV . PEDF—a pigment epithelium-derived factor with potent neuronal differentiative activity. Exp Eye Res 1991; 53: 411–414
Chader GJ . PEDF: raising both hopes and questions in controlling angiogenesis. Proc Natl Acad Sci USA 2001; 98: 2122–2124
Dawson DW, Volpert OV, Gillis P, Crawford SE, Xu HJ, Bendict W, Bouck NP . Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science 1999; 285: 245–248
Spranger J, Osterhoff M, Reimann M, Mohlig M, Ristow M, Francis MK et al. Loss of the anti-angiogenic pigment epithelium-derived factor in patients with angiogenic eye disease. Diabetes 2001; 50: 2641–2645
Stellmach V, Crawford SE, Zhou W, Bouck N . Prevention of ischemia-induced retinopathy by the natural ocular antiangiogenic agent pigment epithelium-derived factor. Proc Nat Acad Sci USA 2001; 98: 2593–2597
Mori K, Duh E, Gehlbach P, Ando A, Takahashi K, Pearlman J et al. Pigment epithelium-derived factor inhibits retinal and choroidal neovascularization. J Cell Physiol 2001; 188: 253–263
Acknowledgements
The authors’ research referred to in this review was funded by The Wellcome Trust and the British Diabetic Association.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cai, J., Boulton, M. The pathogenesis of diabetic retinopathy: old concepts and new questions. Eye 16, 242–260 (2002). https://doi.org/10.1038/sj.eye.6700133
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6700133
Keywords
This article is cited by
-
Lactylation-driven FTO targets CDK2 to aggravate microvascular anomalies in diabetic retinopathy
EMBO Molecular Medicine (2024)
-
Yellow Subthreshold Micropulse Laser in Retinal Diseases: An In-Depth Analysis and Review of the Literature
Ophthalmology and Therapy (2023)
-
A study of the genotyping and vascular endothelial growth factor polymorphism differences in diabetic and diabetic retinopathy patients
Egyptian Journal of Medical Human Genetics (2022)
-
Changes in aqueous and vitreous inflammatory cytokine levels in proliferative diabetic retinopathy: a systematic review and meta-analysis
Eye (2022)
-
Pericyte Biology in the Optic Nerve and Retina
Current Tissue Microenvironment Reports (2022)
