Article Text

Statistics from Altmetric.com
Retinitis pigmentosa (RP) is a generic term used to describe a group of inherited retinal degenerations that lead to the death of rod and cone photoreceptors. RP has a prevalence of about 1:3500. It is clinically and genetically heterogeneous with autosomal dominant, autosomal recessive and X linked inheritance being described.1 Even among these subtypes, there is considerable genetic heterogeneity, with over 100 genes currently known (http://www.sph.uth.tmc.edu/retnet/). One of the challenges for the clinician is how to utilise the results of molecular genetic testing to improve clinical care and our understanding of disease mechanisms. Detailed knowledge of genotype/phenotype correlations allows targeted and therefore cost-effective molecular testing, clearly advantageous in a disorder with so many causative genes, and will help determine which disorders should be prioritised for the development of novel therapies.
Retinitis pigmentosa with preservation of the para-arteriolar retinal pigment epithelium (PPRPE) was first described by Heckenlively et al in 1982.2 In this disorder, there is relative preservation of retinal pigment epithelium (RPE) adjacent to and under retinal arterioles. In 1994, van den Born et al3 reported a clinical study of 22 patients from one large family with PPRPE. Subsequent molecular genetic testing identified the disease locus on 1q31–32.1,4 and den Hollander et al5 subsequently identified CRB1 as the causative gene.
The CRB1 gene is analogous to the Drosophila melanogaster Crumbs protein that is involved in the establishment of the polarity of epithelial cells.6 7 The CRB1 protein is found at the apical region of all retinal epithelial cells, the rod and cone photoreceptor cells and Muller glial cells (MGCs) in the adult mouse retina.8 It colocalises with the zonula adherens (ZA) and is a major component of the outer limiting membrane. Mutations in CRB1 have been identified in a variety of human retinal disorders, including Leber congenital amaurosis (LCA),9–11 PPRPE10 and retinitis pigmentosa.12 13
The aim of this study was to identify mutations in CRB1 in a large cohort of patients and to characterise the ocular phenotype.
Methods
Patient recruitment
Patients with a diagnosis of LCA or early-childhood-onset retinal dystrophy (EORD), and families with autosomal recessive RP where there was parental consanguinity were recruited from two centres: Moorfields Eye Hospital and Great Ormond Street Hospital for Children, London, UK. Ethics committee approval was obtained, and research procedures were carried out in accordance with institutional guidelines and the declaration of Helsinki. Informed consent was obtained for all subjects prior to their participation in the study.
Clinical definitions
All subjects had a family history consistent with AR inheritance. Patients were diagnosed as having LCA if vision was poor or absent in the first 3 months after birth, and where there was no recordable electroretinogram. A diagnosis of early-onset retinal dystrophy was made when symptoms and/or signs were present before the age of 5 years, and subsequently, on electroretinography, there was evidence of a severe cone–rod or rod–cone dystrophy. Children older than 5 years with later onset of symptoms and signs of a retinal dystrophy were termed ‘juvenile onset’ or were labelled synonymously as autosomal recessive retinitis pigmentosa (ARRP).
Clinical evaluation
A full clinical history and examination were performed in all subjects. In older children and adults, this included best-corrected logMAR visual acuity and colour vision testing using the Hardy–Rand–Rittler pseudoisochromatic plates (Richmond Products, Boca Raton, Florida). In the low vision range, for the purposes of later analysis, logMAR values of 2.0 were applied for counting fingers acuity; 3.0 for hand movements, 5.0 for light perception and 6.0 for no light perception.14 Visual-field testing using Goldmann perimetry was performed in those with stable fixation. Subsequent analysis of the visual-field area was performed using planimetry within the V4e isopter.
Refraction data were ascertained either by obtaining an objective cycloplegic refraction using neutralising lenses or using an autorefractor (Luneau L62-3D autorefractokeratometer; Luneau, Chartres, France). Fundus photographs were obtained where possible using the Topcon retinal camera (TRC-50IX with IMAGEnet 2000 system software; Topcon, Tokyo, Japan). Optical coherence tomography scans were performed on a Stratus optical coherence tomograph (OCT) (Carl Zeiss Meditec, Dublin, California); further OCT imaging was performed in some patients using the Topcon 3d OCT-1000 and the Spectralis spectral domain OCT (Heidelberg Engineering, Heidelberg, Germany). Fundus autofluorescence images were obtained using the Heidelberg Retinal Angiograph-II (Heidelberg Engineering). Axial length and keratometry measurements were obtained using the IOL Master (Carl Zeiss Meditec). All patients underwent electrophysiological testing either at our institutions or at the referring hospital: many recordings were historical and either preceded ISCEV standard recordings or incorporated the ISCEV standards pertaining at the time. Where further electrophysiological testing was required to make or confirm a diagnosis, adults and older children had electroretinography conducted in accordance with current ISCEV standards using gold foil or DTL electrodes.15 16 Electroretinogram (ERG) testing for infants and young children in our institutions was performed using skin electrodes in accordance with previously published protocols.17 18
Genotyping
DNA was extracted from whole blood obtained from each affected individual and immediate family members using the Nucleon BACC-2 kit (GE Healthcare, Freiburg, Germany) performed as per the manufacturer’s instructions.
DNA samples from patients with LCA and EORD were sent to Asper Ophthalmics (Tartu, Estonia) for analysis using the LCA chip.19 In those subjects in whom a mutation in CRB1 was identified, DNA samples were then sequenced to confirm the chip findings and, where only one mutation was found, to identify a second change.
DNA from consanguineous families with autosomal recessive RP were analysed using the Affymetrix 100K gene chip. Those showing linkage to the 1q31–32 region were sequenced for CRB1 mutations. DNA samples from patients with recessive RP who had a retinal phenotype characteristic of CRB1 were also sequenced.
Oligonucleotide primer pairs (see appendix 1 published online at http://bjo.bmj.com) were created to cover the exon and splice site junctions (based on RefSeq accession number NM_201253.1) and were designed using Primer 3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) and ordered from Sigma-Genosys (Sigma-Aldrich, St Louis, Missouri). The amplified products were sequenced with Big Dye terminator v1.1 cycle sequencing kits (Applied Biosystems, Warrington, UK) according to the manufacturer’s instructions. The pathogenic impact of the CRB1 sequence variants caused by non-synonymous changes in protein sequence was evaluated through SIFT (http://sift.jcvi.org) and PolyPhen (http://genetics.bwh.harvard.edu/pph/index.html) sequence homology-based programmes.
Results
DNA samples from 250 probands with LCA/EORD were analysed using the LCA chip. Twenty-one probands were found to have mutations in CRB1; seven had two mutations, and 14 had one mutation identified. All mutations were confirmed by direct sequencing, and a second mutation was identified in 11 patients. Two recessive RP families, who were studied with Affymetrix 100k gene chips, showed linkage to the RP12 locus and were found to have homozygous mutations in CRB1. A further 14 RP patients with a clinical phenotype suggestive of CRB1 related disease were also sequenced; nine were found to have mutations in CRB1, and two mutations were identified in six of these patients.
Of the cohort of 306 patients, 41 patients from 32 families were identified as having mutations of CRB1 (table 1). Thirty different sequence variants were identified, two-thirds of which were in exons 7 and 9. Seventeen novel disease associated sequence variants were found. None of these variants were found in 100 normal controls, and appropriate segregation of alleles in family members was confirmed where DNA was available. The most common mutation identified was the p.C948Y variant, which was present in 13 alleles of 11 patients. Six probands had one mutation found, and in 26 probands both alleles were identified. Of the latter group, seven had homozygous mutations, and 19 had compound heterozygous mutations. Mutations in the CRB1 gene were identified in 7% of the LCA/EORD cohort.
Results of CRB1 mutation screening in Leber congenital amaurosis (LCA), early-onset rod–cone dystrophy (EORCD), early-onset cone–rod dystrophy (EOCRD) and autosomal recessive retinitis pigmentosa (ARRP) patients
Clinical findings
Thirty-four patients from 26 families had two CRB1 mutations identified. They ranged in age from 2 to 48 years (mean 19 years; 22 male, 12 female). Twenty-one patients were Caucasian, 11 were from the Indian subcontinent, and two were African. The median visual acuity for the whole cohort was 1.6 logMAR. The median and range of logMAR visual acuity measurements for each decade are given in table 2.
Cross-sectional analysis of the median and range of logMAR visual acuity measurements in all patients with mutations in CRB1
The median spherical equivalent refraction for all patient measured was +1.5 dioptres (D). Funduscopy revealed the majority of patients to have nummular pigmentation at the level of the retinal pigment epithelium that was more widespread in the older patients. Ten patients (29%) had macular atrophy, and six (18%) had PPRPE. Five (15%) subjects had peripheral retinal telangiectasia, which in three cases was associated with exudative retinal detachment. Two patients had seclusio pupillae and secondary glaucoma. The median foveal thickness on Stratus OCT was 326 μm (range 212–854 μm; SD 148 μm).
The LCA patients all had a non-detectable ERG. Visual acuity ranged from 0.9 logMAR to no perception of light. Refraction ranged from +4.5 to +9.5 D (spherical equivalent) and correlated with short axial length, which ranged from 17.95 mm to 23.06 mm (table 3). Colour vision was absent in all but one patient, who was able to discern the highest-contrast HRR plates. Reliable OCT measurements could only be made in six patients because of nystagmus; foveal thickness ranged from 212 μm to 854 μm with intraretinal cystic spaces seen in two patients. Funduscopy showed white dots at the level of the RPE in the younger patients and deep nummular pigment in older subjects (figure 1D). Macular atrophy was present in seven of the 15 (46%) patients with LCA (figure 1F). Analysis of mutations in this group of patients showed that 23/24 alleles were either protein-truncating (9/24)—both nonsense and frame-shifting—or predicted by both SIFT and PolyPhen analysis to severely affect protein function (14/24).
Median logMAR visual acuity, refraction, visual field size, axial length and foveal thickness, and number of patients with retinal signs by diagnosis

Fundus images and Topcon 3d optical coherence tomography in patients with mutations in CRB1. (A) Patient 38 with early-onset childhood retinal dystrophy and bilateral peripheral telangiectasia with extensive subretinal exudates. (B) Patient 37 with cone–rod retinal dystrophy; the fundus abnormalities are confined to the macula. (C) Patient 13 illustrating para-arteriolar retinal pigment epithelium with early onset rod–cone dystrophy (EORCD). (D) Patient 16 with Leber congenital amaurosis (LCA) and widespread nummular pigmentation. (E) Patient 18 with EORCD; Topcon 3d optical coherence tomography shows thickening of macula with preservation of foveal architecture and some preservation of retinal laminae. (F) Patient 22 with LCA; the fundus image shows macular atrophy, white dots in retinal mid-periphery and peripheral nummular pigmentation.
The early-onset rod–cone dystrophy patients were between 5 and 39 years of age when examined. Nine patients had a historical diagnosis of ARRP but, on further questioning, had symptoms that were childhood or juvenile in onset. The median onset of symptoms was at 2.5 years (range 6 months to 5 years). The median age at diagnosis was 6 years. Visual acuities ranged from 0.16 logMAR to perception of light. The spherical equivalent refractive error ranged from −0.5 D to +6.5 D with the majority having low hyperopia. Axial lengths ranged from 19.03 mm to 23.35 mm. The foveal thickness measurements, on Stratus OCT, ranged from 217 to 396 μm. Intraretinal cysts were common (figure 2B–D). The visual field was relatively well preserved in several patients, and the field area ranged from 102 to 10 000 square degrees. Fundus examination revealed widespread pigment clumping at the level of the RPE in the majority of cases. One patient exhibited PPRPE (figure 1C).

Spectralis images of (A) normal retina illustrating laminar arrangement. NFL, nerve fibre layer; GCL, ganglion cell layer; IPL, inner plexiform layer; OPL, outer plexiform layer; ONL, outer nuclear layer; OLM, outer limiting membrane; PRIS, photoreceptor inner segment; IS/OS, inner segment/outer segment boundary; PROS, photoreceptor outer segment; RPE, retinal pigment epithelium; BM, Bruch's membrane; CC, choriocapillaris. (B–D) Siblings 13–15: (B) age 9 years, (C) age 18 years, (D) age 22 years with early-onset childhood retinal dystrophy and homozygous C250W mutation. Progressive photoreceptor and retinal pigment epithelium loss with age is observed, with pigment migration to nerve-fibre layer. Cystic cavities within the inner and outer nuclear layers were noted in all patients. The OLM is preserved in patients B and C; in patient D there is loss of the OLM, and the number of discernible laminae is reduced. (E) Patient 38 with early-onset childhood retinal dystrophy and extensive intraretinal exudation secondary to peripheral retinal telangiectasia; the photoreceptor layer is absent, but coarse retinal laminae can still be observed.
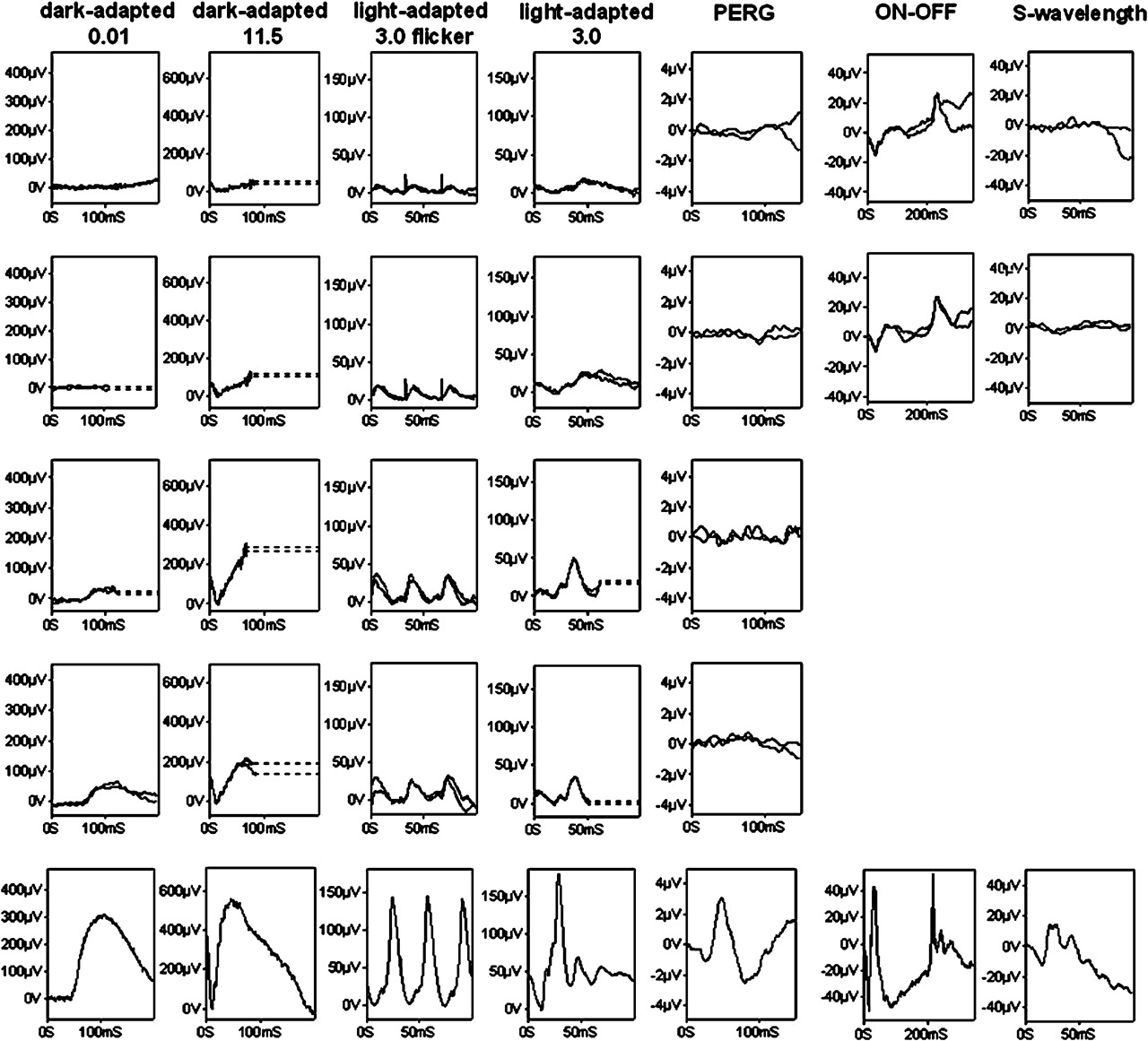
The three patients with cone–rod dystrophy were aged 13, 17 and 19 years at examination. Poor central vision was the first symptom with later development of night blindness and visual-field loss. One patient had peripheral retinal telangiectasia and exudative retinal detachment (figures 1A, 2E). The ERG findings suggested generalised photoreceptor dysfunction with evidence of more severe macular involvement (figure 3).
{kind=link}
{kind=link}
{kind=link}
International-standard full-field electroretinograms (ERGs) and pattern ERGs 14, 15 from the right eyes of patients 37 (row 1), 17 (row 2), 36 (row 3), 14 (row 4) and typical normal examples for comparison (row 5). The electrophysiological findings are consistent with cone–rod dystrophy with severe macular dysfunction (rows 1 and 3), severe generalised photoreceptor dystrophy with severe macular dysfunction (row 2) or rod–cone dystrophy with some sparing of central macular function (row 4). Dark-adapted ERGs are shown for flash intensities of 0.01 and 11.5 cd.s/m2; light-adapted ERGs for a flash intensity of 3.0 cd.s/m2. On–off ERGs used an orange stimulus (560 cd/m2, duration 200 ms) superimposed on a green background (150 cd/m2). Short-wavelength ERGs used a blue stimulus (445 nm, 80 cd/m2) on an orange background (620 nm, 560 cd/m2). Broken lines replace eye-movement artefacts for clarity. PERG, pattern electroretinogram.
Examination of the mutations in these two groups of early-onset patients showed that 16/18 alleles would be predicted to affect protein function severely, similar to the pattern seen in the LCA group. Three of 18 alleles contained nonsense mutations; 13/18 alleles contained mis-sense mutations predicted to have a severe effect on protein function.
The patients with juvenile onset rod–cone dystrophy had a less severe phenotype and later onset of disease. The median age at diagnosis was 10 years. Visual acuities ranged from 0.28 logMAR to no perception of light, with the median acuity 0.8 logMAR. Low hypermetropia was noted in all but one patient. Visual-field planimetry ranged in size from 620 to 11 160 square degrees. Foveal thickness, measured by OCT, ranged from 322 to 544 μm, with several patients exhibiting intraretinal cysts. The retinal appearances were more heterogeneous and ranged from a grey discoloration of the RPE around the arcades with PPRPE to extensive retinal pigmentation. Retinal telangiectasia was seen in two patients who subsequently developed retinal detachment, uveitis with seclusio pupillae and angle-closure glaucoma. In this group of patients, there was a lower prevalence of protein truncating mutations and a higher prevalence of missense mutations compared with those subjects with LCA.
Discussion
The present study investigated the incidence of CRB1 mutations in a large cohort of patients with recessive retinal dystrophies reports 17 novel mutations. An APEX microarray was used as an initial screening strategy, and CRB1 mutations were identified in 7% of patients with LCA and EORD. This is likely to be an underestimate, as sequencing of CRB1 was not performed in all cases. When cases with a suggestive clinical phenotype were also included, the detection rate in this cohort increased to 12%, indicating that CRB1 mutations are a common cause of LCA and childhood-onset rod–cone dystrophies.
Mutations in CRB1 account for 10–13% of patients with LCA,9–11 but CRB1 mutations have also been identified in patients with autosomal recessive retinitis pigmentosa (RP12)5 20 where there is often para-arteriolar sparing of the RPE. A CRB1 mutation has also been reported in a single family with autosomal dominant pigmented paravenous chorioretinal atrophy (PPCRA) that exhibited variable expressivity.21 Peripheral retinal telangiectasia and associated exudation is more commonly associated with CRB1 mutations than other forms of RP.3 13 We observed telangiectasis, exudation and subsequent development of retinal detachment in 15% of our cohort. It seems plausible that this ‘Coats-like’ exudative vasculopathy is secondary to a breakdown in the normal blood retinal barrier mediated by loss of normal zonula occludens function and therefore immune exposure to retinal antigen.22 The characteristic appearances of exudation and retinal detachment, yellow lipid deposition, with subsequent uveitis and neovascular glaucoma would seem to be a final common pathway of abnormal retinal development whether the cause is secondary to mutations in CRB1, Norrin or associated with FEVR and Coats disease.
We identified CRB1 mutations in patients with LCA, early-onset rod–cone dystrophy, juvenile-onset RP, and cone–rod dystrophy. The severity of the retinal disease generally correlated with the age of onset of symptoms. The disease appears clinically progressive, though longitudinal data were not collected; however, all patients examined who were in their fourth decade had visual acuities of counting fingers or worse. There is a readily recognisable phenotype that can help guide molecular genetic testing. In infants, there are typically widespread subretinal white dots, and macular atrophy is common. The characteristic deep nummular pigmentation usually develops in later childhood, and the pigmentation becomes more extensive with age. PPRPE, present in 18% of our patients, is strongly suggestive of CRB1-related disease, particularly when accompanied by peripheral telangiectasia. A variety of macular changes were found: macular atrophy and loss of central macular volume on OCT were noted, particularly in patients with LCA and early-onset rod–cone dystrophy. No clear relationship with genotype was established, though it was noted that six of the 13 patients in the cohort that had either heterozygous or homozygous frame-shifting/termination mutations had macular atrophy noted on funduscopy.
OCT imaging is also extremely helpful in identifying CRB1-related disease. Jacobson et al23 first described increased retinal thickness and loss of the normal retinal laminae in patients with CRB1 mutations. All of the patients in the current study who had OCT showed significantly increased retinal thickness. Newer spectral domain OCT imaging permits better visualisation of the laminar arrangement of the inner retina. In one family, with three affected individuals, spectral domain OCT showed well-delineated retinal layers with preservation of the outer limiting membrane (OLM) in the younger patients but coarse lamination and loss of the OLM in the oldest subject (figure 2B–D). This suggests that the coarse lamination seen on OCT may not represent a developmental abnormality, but that loss of the normal architecture develops with time. The CRB1 protein is known to play a critical role in retinal development6 8 24 and patients with CRB1 mutations have abnormal retinal structure as a consequence. Longitudinal studies are required to determine whether there is progressive loss of the OLM, and what role this may have in determining the increased retinal volume seen on OCT.
The preliminary results of gene-replacement therapy for LCA associated with mutations in RPE65 have been promising,25–27 and this has led to optimism that similar treatment strategies may be helpful in other forms of LCA and childhood retinal dystrophy. The retinal disease associated with CRB1 mutations is generally more severe, but there are some patients in whom there is a window of opportunity for therapy in early childhood.
References
Footnotes
Funding Fight for Sight, British Retinitis Pigmentosa Society, Ulverscroft foundation, Foundation Fighting Blindness USA, National Institute for Health Research (NIHR—Moorfields Biomedical Research Centre).
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was provided by the Moorfields Research Ethics Committee Approval Number (MOA1005).
Provenance and peer review Not commissioned; externally peer reviewed.