Article Text

Abstract
Aim The current accepted standard treatment for neovascular age-related macular degeneration (AMD) consists of antivascular endothelial growth factor agents including ranibizumab and bevacizumab. The aim of the study was to examine whether bevacizumab is inferior to ranibizumab with respect to maintaining/improving visual acuity.
Methods In this prospective randomised parallel group multicentre trial patients aged more than 50 years with treatment naive nAMD were included at 10 Austrian centres. Patients were randomised to treatment either with 0.5 mg ranibizumab or 1.25 mg bevacizumab. Both groups received three initial monthly injections and thereafter monthly evaluation of visual acuity and the activity of the lesion. Re-treatment was scheduled as needed. Outcome measures were early treatment of diabetic retinopathy visual acuity, retinal thickness, lesion size and safety evaluation.
Results A total of 321 patients were recruited of which four had to be excluded due to different reasons. Of the 317 remaining patients 154 were randomised into the bevacizumab group and 163 into the ranibizumab group. At month 12, there was a mean increase of early treatment of diabetic retinopathy visual acuity of 4.9 letters in the bevacizumab and 4.1 letters in the ranibizumab group (p=0.78). Furthermore, there were no significant differences in the decrease of retinal thickness, change of lesion size and number of adverse events between the groups.
Conclusions Bevacizumab was equivalent to ranibizumab for visual acuity at all time points over 1 year. There was no significant difference of decrease of retinal thickness or number of adverse events.
- Retina
- Treatment Surgery
- Neovascularisation
- Macula
Statistics from Altmetric.com
Introduction
The healthcare systems of the Western world are confronted with an increasing number of patients suffering from age-related macular degeneration (AMD).1 Friedman et al2 estimated an increase of 50% of the number of individuals suffering from AMD in the USA up to the year 2020. Although the neovascular form of AMD accounts for only 20% of the cases with AMD, it is responsible for 90% of the cases of legal blindness.1 In recent years, significant advances in the treatment of neovascular AMD have been achieved. In contrast to prior treatments such as argon laser photocoagulation and photodynamic therapy (PDT), modern approaches with inhibitors of anti-VEGF (vascular endothelial growth factor) are not limited by lesion composition and lesion size.3 ,4 Treatment with the anti-VEGF agents ranibizumab and bevacizumab has become the mainstay of the treatment of neovascular AMD. Ranibizumab (Lucentis) was approved in this indication by the Food and Drug Administration and by the European Medicines Agency. Bevacizumab (Avastin) was approved for the therapy of colorectal cancer but is frequently used off label for intravitreal injection. The active part of the antibody is similar in ranibizumab and bevacizumab. However, there are a series of differences with a possible impact on safety and effect of the treatment. The antibody fragment ranibizumab is smaller (48 kD) than the whole anti-VEGF bevacizumab (150 kD). In comparison with bevacizumab, ranibizumab provides a shorter half-lifetime, lower serum concentrations, a better penetration through the retina and a several-fold binding to VEGF-A.5–7 Furthermore, the costs of one injection of ranibizumab may reach 50 times that of bevacizumab.8 ,9
Large multicentre studies have proven that monthly applied ranibizumab is effective and safe in the treatment of neovascular AMD.10 ,11 In clinical practice, frequently as needed regimen (pro re nata (PRN)) was applied, which has been proven to be effective.12 For bevacizumab, a series of smaller studies with a lower evidence level have shown the efficacy of the therapy, most commonly applied in a PRN regimen13–15 and two recent multicentre studies have shown non-inferiority over ranibizumab.16 ,17
Methods
In this prospective randomised double masked parallel group study performed at 10 clinical ophthalmological centres in Austria, a total of 321 patients were included. Consecutive treatment naive patients aged over 50 years with active primary or recurrent subfoveal lesion with choroidal neovascularisation (CNV) secondary to AMD were included. Activity was proven by fluorescein angiography as described previously.10 Furthermore, the optical coherence tomography (OCT) scans were evaluated for intraretinal or subretinal fluid. Best corrected visual acuity (BCVA) in the study eye assessed using early treatment of diabetic retinopathy (ETDRS) charts had to be 20/40 to 20/320. The exclusion criteria for patients in the present trial are presented in box 1.
Exclusion criteria
-
Prior treatment with any intravitreal drug in the study eye
-
Prior treatment with verteporfin photodynamic therapy in the study eye
-
Prior treatment with systemic bevacizumab
-
Prior treatment with any intravitreal drug or verteporfin photodynamic therapy in the non-study eye within the 3 months before the study entry
-
Laser photocoagulation within 1 month before study entry in the study eye
-
Previous participation in any clinical trial within 1 month before the entry of the study
-
Subfoveal fibrosis or atrophy in the study eye >50%
-
Choroidal neovascularisation in either of the two eyes due to causes other than age-related macular degeneration such as histoplasmosis or pathological myopia (refractive error in the study eye demonstrating more than –6 dioptres or an axial length of ≥26 mm of myopia)
-
Retinal pigment epithelial tear involving the macula in the study eye
-
History of uncontrolled glaucoma in the study eye (defined as intraocular pressure ≥25 mm Hg despite treatment with topical antiglaucomatous medication)
-
Any concurrent intraocular condition in the study eye that could either require medical or surgical intervention during the 12-month study period or that could contribute to a loss of best corrected visual acuity over the 12-month study period (eg, diabetic retinopathy, cataract, uncontrolled glaucoma). The decision on exclusion is to be based on the opinion of the local principal investigator
-
Active intraocular inflammation
-
Vitreous haemorrhage in the study eye
-
Pregnancy (a pregnancy test will be done monthly in women of childbearing potential)
-
History of rhegmatogenous retinal detachment or stage 3 or 4 macula hole in the study eye
-
History of idiopathic or autoimmune-associated uveitis in either eye
-
Acute or recurrent infectious conjunctivitis, keratitis, scleritis or endophthalmitis in either eye
-
Aphakia or absence of the posterior capsule in the study eye
-
Intraocular surgery in the study eye within 2 months before the entry of the study
-
History of corneal transplant in the study eye
-
History of myocardial infarction and/or stroke
-
History of any ocular or systemic disease that according to the opinion of the local principal investigator may affect the interpretation of the study results or render the subject at high risk for treatment complications including severe hypertension
-
History of allergy to fluorescein, not amendable with diphenhydramine
-
Inability to comply with study procedures
If both eyes were eligible for inclusion in the present study, the eye that showed more progression (loss of distance acuity) based on the local investigator's assessment was included. The non-study eye of the patient's was treated independently of the aims of the present study. Eligible patients were randomised in a 1:1 ratio to one of two groups by members of the Department of Clinical Pharmacology, Medical University of Vienna, which was otherwise not involved in the study. Randomisation was stratified according to the clinical centre using a permuted block method with a fixed block size of 20. Group 1 received intravitreal injections of 0.5 mg ranibizumab (Lucentis, Novartis, Basel, Switzerland), and group 2 received intravitreal injections of 1.25 mg bevacizumab (Avastin, Genentech, San Francisco, USA). Both groups received 3 monthly intravitreal injections, thereafter re-injections were scheduled if any of the following changes was observed at a study visit: (1) visual acuity loss of at least five letters with OCT or fluorescein angiographic evidence of fluid in the macula; (2) an increase in OCT central retinal thickness of at least 100 μm; (3) new macular haemorrhage; (4) new area of classic CNV; or (5) evidence of persistent fluid on OCT at least 1 month after the previous injection. Antimicrobial drops were administered before and after intravitreal injection according to the standards at each participating site. Antiseptic preparation and pretreatment with local anaesthesia were performed as usual.18 Study drugs were prepared by the local pharmacies and stored refrigerated until use. Masking to treatment required at least two investigators per site. The evaluating physician was masked to treatment assignment, whereas the injecting physician was not involved in the collection of data. All other personnel and the patients were masked to treatment assignment.
The main outcome variable was the mean change in BCVA between baseline and 1 year. Secondary outcomes included: (1) Kaplan–Meier proportions of the gain of 15 letters of vision, (2) Kaplan–Meier proportions of the gain of five letters of vision, (3) Kaplan–Meier proportions of the loss of five letters of vision, (4) Kaplan–Meier proportions of the loss of 15 letters of vision, (5) Lesion size assessed by fluorescein angiography, (6) Number of re-treatments, (7) Retinal thickness (OCT) and (8) Adverse events.
Examinations
Study visits were scheduled at screening, baseline and thereafter monthly (30±7 days). Each visit consisted of:
-
Biomicroscopy of the anterior and posterior segment
-
Assessment of intraocular pressure
-
BCVA
-
OCT
-
Concomitant medication
-
Assessment of adverse events (monthly exploration of the patients and documentation in the case record forms).
Fluorescein angiography was performed at the screening and month 12 visit. BCVA was tested with ETDRS charts at 4 m distance. For OCT examinations, either Stratus OCT or Cirrus OCT or Spectralis OCT could be used; central retinal thickness (mean value of retinal thickness within the central area with a diameter of 100 µm) values were converted as previously described.19 ,20
Sample size calculations
The number of 320 patients was calculated based on the distance acuity results of three randomised clinical trials with ranibizumab.10–21 The main outcome parameter for the present trial was chosen as mean change in visual acuity over time. In the previous trials, the increase in visual acuity was approximately seven letters with ranibizumab. The present sample size was calculated based on a 95% power to detect a significant difference between ranibizumab and bevacizumab assuming a seven letters increase in visual acuity with ranibizumab and no change in visual acuity with bevacizumab. This is considered the minimal clinically relevant difference. The study was not powered to determine adverse events of statistical significance.
Biometric methods
Efficacy analysis was performed on an intention to treat basis for all randomised patients using a last observation carried forward method to handle missing data. For all pair-wise comparisons, the model was stratified by baseline visual acuity scores. Cochran λ2 was employed for between group comparisons for dichotomous endpoints. For all other outcomes, analysis of covariance models adjusted for baseline values were used. Kaplan–Meier survival curves were calculated using Cox's proportional hazard model with time-dependent covariates (Statsitica V.7.0, StatSoft, Inc. Tulsa, Oklahoma, USA).
Results
Out of 321 randomised patients four patients were excluded. In three patients the wrong drug was injected into the study eye. In one patient the drug was administered although the patient was not treatment naive. The baseline data of the study groups are presented in table 1. No major differences were seen between the patients receiving ranibizumab and those receiving bevacizumab. Out of the 154 patients in the bevacizumab group, 121 visual acuity scores were available after 1 year (78.6%). In the ranibizumab group, 127 visual acuity scores were available after 1 year from the 163 included patients (77.9%).
Baseline characteristics of the study population, data are presented as mean±SD
Visual acuity increased in both study groups after 1 year. The visual acuity was 60.7 letters (CI 58.7 to 62.8 letters) in the ranibizumab group and 62.2 letters (CI 60.1 to 64.3 letters) in the bevacizumab group after 12 months. An improvement in BCVA was already seen after 1 month and reached its maximum 3–4 months after initiation of treatment. Figure 1 shows the time course of BCVA over the 12-month study period. No significant difference was found between ranibizumab and bevacizumab (p=0.78 between groups). At 1 year, bevacizumab was equivalent to ranibizumab (CI for the difference in the mean change in BCVA within −7 to +7 letters). The two drugs were also not significantly different when results were adjusted for age and baseline BCVA.

Effect of bevacizumab (n=154, blue) versus ranibizumab (n=163, red) on the change in best corrected visual acuity (BCVA). Data are presented as mean±95% CI.
The proportion of patients gaining at least five or 15 letters during treatment is shown in figure 2. No significant difference was found between the study groups, neither for the five letters margin (p=0.31) nor for the 15 letters margin (p=0.42). The proportion of patients with a gain in BCVA tended to be higher initially in the ranibizumab group, but later tended to be lower than in the bevacizumab group. The number of patients losing at least five or 15 letters during treatment is shown in figure 2. Again there was no significant difference in the treatment groups between the two drugs (five letters margin: p=0.11; 15 letters margin p=0.23).
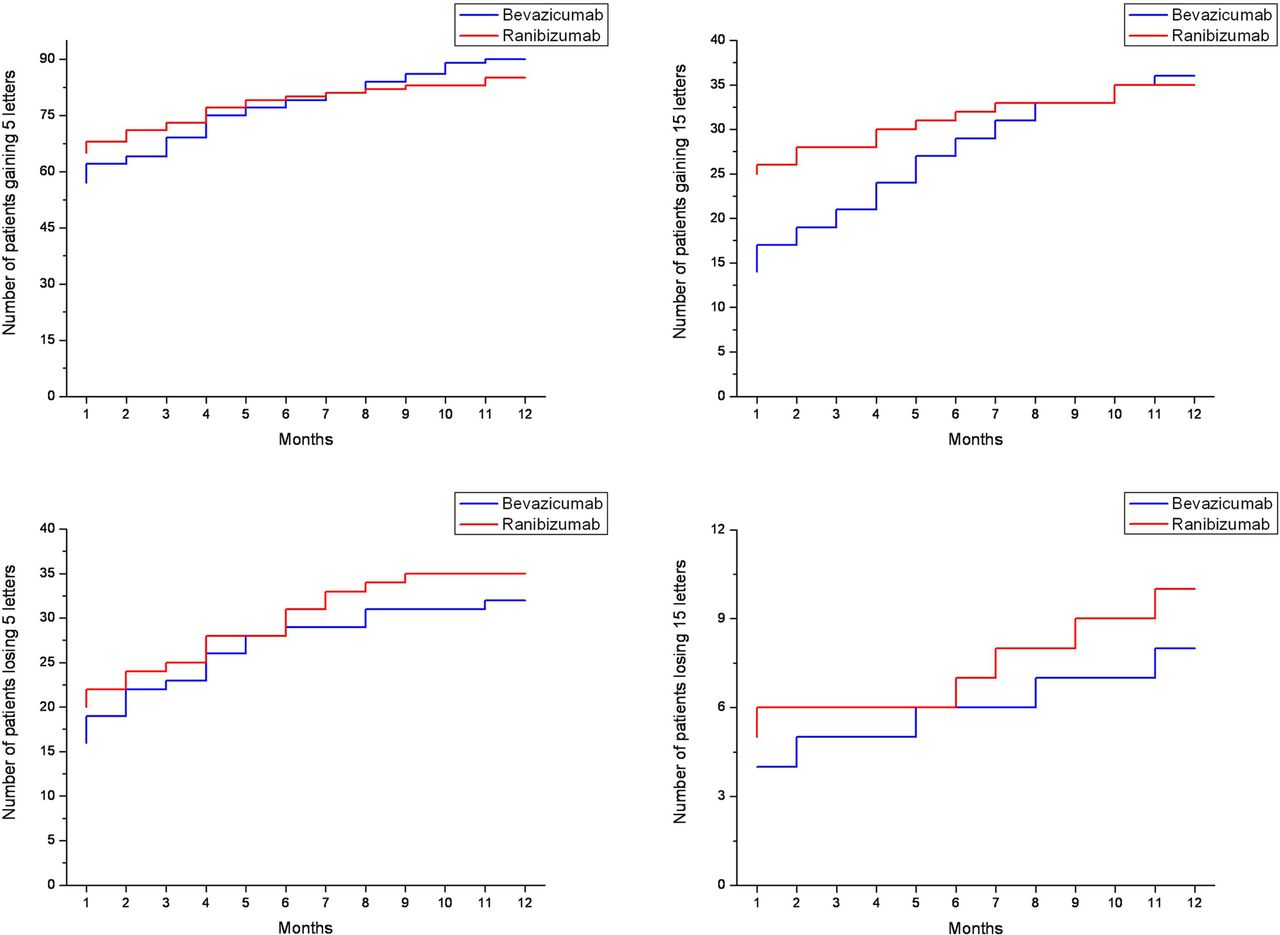
Upper panel: Kaplan–Meier curves presenting number of patients gaining five letters (left) or more and gaining 15 letters or more (right) in the bevacizumab (n=154, blue) and ranibizumab (n=163, red) groups over time. Lower panel: Kaplan–Meier curves presenting number of patients losing five letters or more (left) or 15 letters or more (right) in the bevacizumab (n=154, blue) and ranibizumab (n=163, red) groups over time.
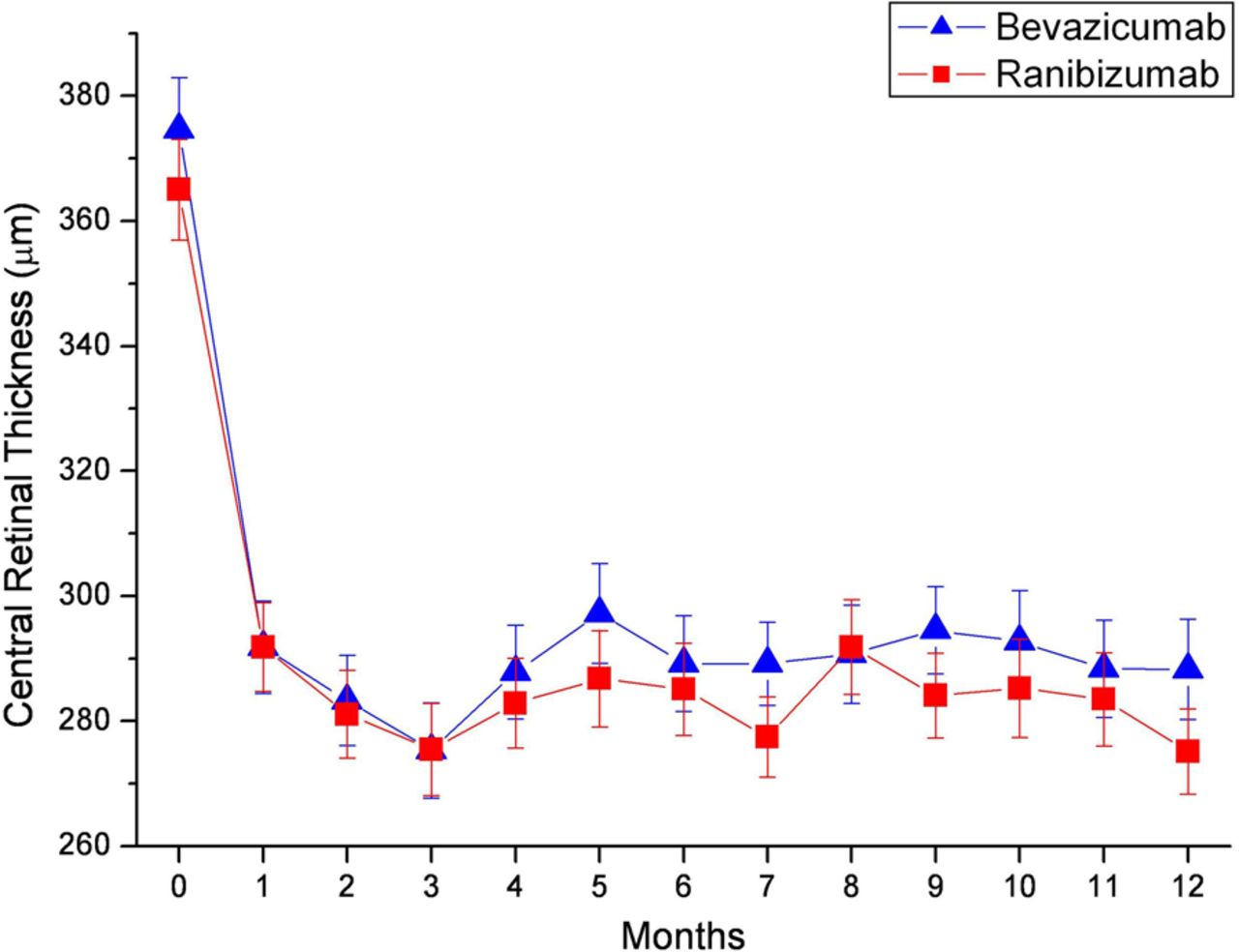
In all, 37.6% of OCT examinations were performed with Cirrus OCT (Carl Zeiss, Dublin, USA), 34.3% with Spectralis OCT (Heidelberger engineering, Heidelberg Germany) and 28.1% with Stratus OCT (Carl Zeiss, Dublin, USA). The central retinal thickness corrected values (converted to compensate for different OCT machines used19 ,20) decreased from 374.59 µm±8.4 to 288.29 µm±8.0 in the bevacizumab group and from 365.0 µm±8.1 to 275.14 µm±6.8 in the ranibizumab group. The differences were not significant between the groups (p=0.81). The course of the decrease of retinal thickness is presented in figure 3.
{kind=link}
{kind=link}
{kind=link}
Effect of bevacizumab (n=154, blue) versus ranibizumab (n=163, red) on the change of retinal thickness over time. Data are presented as mean±95% CI.
The mean number of re-treatments in the study was 5.8±2.7 in the ranibizumab group and 6.1±2.8 in the bevacizumab group (p=0.26). During the observation period, six patients required treatment also in the fellow eye, none of them at baseline (four in the ranibizumab, two in the bevacizumab group); the patients received the same agent as was randomised for the study eye. No significant difference was observed in terms of lesion size between the two groups (p=0.55). Mean lesion size decreased from 3.02±0.14 disk areas in the bevacizumab group and 2.85±0.13 in the ranibizumab group to 0.68±0.16 and 0.63±0.14, respectively.
Table 2 summarises the frequency of serious adverse events in the study. Neither the total number of adverse events nor the number of adverse events in any of the subgroups was significantly different between the groups.
Frequency of adverse events
Discussion
Recently, the 12-month results of the first two head-to-head studies became available, the Comparison of Age-related Macular Degeneration Treatment Trials (CATT)16 conducted in the USA and the Alternative Treatments to Inhibit VEGF in Age-related Choroidal Neovascularisation Trial (IVAN)17 conducted in the UK. In contrast to these trials, which compared a continuous with a discontinuous treatment regimen for both substances, the MANTA Study concentrated on a treatment regimen most frequently applied in clinical practice, namely PRN. After initial three injections, patients were treated when there were signs of persistent activity in the OCT, furthermore when there was a loss of BCVA of more than five letters or signs of new CNV or blood. Evaluation and treatment (as needed) were performed monthly (30±7 days) similar to the treatment regimen of the discontinuous part of the CATT study but different from the IVAN study (when activity was seen 3-monthly injections were applied).
In the MANTA study, the main outcome measure was the change of BCVA after 1 year using the ETDRS protocol. Mean baseline distance acuity was comparable (57.0 and 56.4 letters), and both groups gained improvement of BCVA, the bevacizumab group 4.9 letters and the ranibizumab group 4.1 letters. This was less than in the CATT study (5.9 and 6.8 letters, respectively, baseline letter score 61.5 and 60.4) and seemed to be equal to the IVAN study (change of letters was not provided). In the MANTA study, a non-inferiority approach was chosen like in the other head to head studies with a limit of seven letters (CATT: five letters; IVAN: three letters) and like in the other studies both drugs showed a similar effect on BCVA. In both groups there was a steep increase over the first 3–4 months; thereafter, distance acuity could be maintained. This initial increase tended to be more prominent in the ranibizumab group and was equalised in the later course. However, these changes were not conclusive. This increase of BCVA was achieved by mean 6.1 and 5.8 treatments for bevacizumab and ranibizumab, respectively. In the PRONTO study,12 a comparable number of treatments was required (5.6), and an analysis of the trends of treatment in clinical practice reported a mean number of 4.3 injections per treated eye.22 In the CATT study mean 7.7 and 6.9 injections, respectively, were applied (re-treatment, when active CNV was present every 28 days). This outcome difference might be explained by the fact that in the MANTA trial an inclusion criterion was central involvement of the CNV, whereas in the CATT study the baseline CNV involved the centre in only 58% of the cases.
A series of OCT machines are on the market. Besides the Stratus OCT (time domain technology), different machines with modern spectral domain technology are used in Austria. To give all the larger ophthalmological departments in Austria the opportunity to participate, different OCT machines were accepted and the resulting different retinal thickness values were converted.19 ,20 The decrease of retinal thickness was comparable for both drugs, as it was seen in the CATT and IVAN trials. Absolute values and differences are not comparable between the MANTA study and the CATT (−152.11 µm in the bevacizumab and −168.11 µm in the ranibizumab groups) and IVAN trials (differences in the discontinuous part separately for bevacizumab and ranibizumab not presented), because in the Austrian study mean values of the central area were given and single point measurements in CATT and IVAN trials. Furthermore, the change of lesion size did not reveal significant differences between the groups.
The systemic reviews reported a low frequency of severe adverse events for both substances.23 ,24 Van der Reis et al24 reviewed 278 articles and found a cumulative incidence of severe adverse events below 1% for both drugs (non-randomised studies and case reports also included). Similarly in the MANTA study the frequency of adverse events was overall low, with slightly more adverse events in the bevacizumab group than in the ranibizumab group (12.3% vs 9.2%), but the differences did not reach statistical significance. These results were comparable with those found in the IVAN trial (12.5% vs 9.6%). In the CATT trial overall higher numbers of adverse events for both groups were reported, again with a tendency towards more adverse events in the bevacizumab group (25.7% vs 20.5%). Overall five patients died (1.9% in the bevacizumab, one heart attack, one mesenteric artery occlusion, one carcinoma of the pancreas; 1.2% in the ranibizumab group, one heart attack, one stroke) in the present study. Severe vascular disorders were reported in 3.2% with bevacizumab and 1.9% with ranibizumab respectively in our study population similar to the observations in CATT (3.0% and 2.7% bevacizumab vs ranibizumab) and IVAN (1.4% and 2.9% bevacizumab vs ranibizumab). In contrast to the CATT study there were no cases of endophthalmitis or pseudoendophthalmitis in our study. Whether this is related by the fact that treatments in the MANTA study were exclusively performed in large ophthalmological departments in surgical settings needs further evaluation.
To summarise, bevacizumab and ranibizumab provided comparable results concerning BCVA, retinal thickness, lesion size and number of adverse events during 1 year.
Acknowledgments
The authors appreciate the efforts of the coworkers in the participating centres: Department of Ophthalmology Rudolf Foundation Clinic (Professor Dr Susanne Binder, Dr Ilse Krebs); Department of Ophthalmology Hospital Barmherzige Brüder Linz (Professor Dr Ulrich Schönherr, Dr Karl Haas); Department of Ophthalmology Medical University Salzburg (Professor Dr Günther Grabner, Professor Dr Stefan Egger); Department of Ophthalmology Medical University Graz (Professor Dr Andreas Wedrich, Professor Dr Anton Haas, Professor Dr Martin Weger, Dr Iris Steinbrugger); Department of Ophthalmology Hospital Hietzing (Dr Veronika Vecsei-Marlovits, Dr Ramin Baraderan-Dilmaghani); Department of Ophthalmology Hanuschkrankenhaus Vienna (Professor Dr Oliver Findl, Dr Stephan Radda); Department of Ophthalmology Medical University Innsbruck (Professor Dr Nikolaos Bechrakis, Professor Dr Gerhard F Kieselbach; Department of Ophthalmology Medical University Vienna (Professor Dr Ursula Schmidt-Erfurth); Department of Ophthalmology Hospital Barmherzige Brüder Vienna (Professor Dr Michael Amon, Dr Walter Steindl); and Department of Ophthalmology General Hospital Linz (Professor Dr Siegfried Priglinger).
References
Footnotes
Contributors IK: substantial contribution to conception and design, acquisition of data, analysis and interpretation of data, drafting the article, final approval. LS: substantial contribution to conception and design, acquisition of data, analysis and interpretation of data, monitoring, revising the article, final approval. AB and RT: substantial contribution to acquisition of data, analysis of data, revising the article, final approval. VV-M, SE, US, AH and SA-S: substantial contribution to conception and design and acquisition of data, revising the article, final approval. SB: substantial contribution to conception and design, revising the article, final approval, general supervision of the research group.
Funding The study was supported by the Austrian ophthalmologic society, by the Ludwig Boltzmann Institute of Retinology and Biomicroscopic Lasersurgery, and by the participating study centres themselves.
Competing interests None.
Patient consent Obtained.
Ethics approval The study was approved by the institutional review boards and by the Austrian National Institute of Health. The trial was registered with the number NCT00710229. Every patient signed a written consent and the data collection was adherent to the tenants of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed .