Article Text

Abstract
Aim To assess the efficacy and safety of latanoprostene bunod (LBN) compared with latanoprost 0.005%, and to determine the optimum drug concentration(s) of LBN in reducing intraocular pressure (IOP) in subjects with open angle glaucoma or ocular hypertension.
Methods Randomised, investigator-masked, parallel-group, dose-ranging study. Subjects instilled one drop of study medication in the study eye once daily each evening for 28 days and completed five study visits. The primary efficacy endpoint was the reduction in mean diurnal IOP at Day 28.
Results Of the 413 subjects randomised (LBN 0.006%, n=82; LBN 0.012%, n=85; LBN 0.024%, n=83; LBN 0.040%, n=81; latanoprost, n=82), 396 subjects completed the study. Efficacy for LBN was dose-dependent reaching a plateau at 0.024%–0.040%. LBN 0.024% led to significantly greater reductions in diurnal IOP compared with latanoprost at the primary endpoint, Day 28 (p=0.005), as well as Days 7 (p=0.033) and 14 (p=0.015). The incidence of adverse events, mostly mild and transient, was numerically higher in the LBN treatment groups compared with the latanoprost group. Hyperaemia was similar across treatments.
Conclusions LBN 0.024% dosed once daily was the lower of the two most effective concentrations evaluated, with significantly greater IOP lowering and comparable side effects relative to latanoprost 0.005%. LBN dosed once daily for 28 days was well tolerated.
Clinical trial number NCT01223378.
- Clinical Trial
- Drugs
- Glaucoma
- Intraocular pressure
- Treatment Medical
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Glaucoma is a leading cause of blindness worldwide.1–3 Intraocular pressure (IOP) is the primary risk factor for glaucoma,3 ,4 and lowering IOP to prevent optic nerve injury is the only proven effective treatment.3 ,5–9 Topical prostaglandin analogues, such as latanoprost, are considered first line treatment to lower IOP due to their safety and efficacy.10–12 BOL-303259-X, also known as latanoprostene bunod (LBN), is a new IOP-lowering agent that, when exposed to ubiquitous esterases in the ocular environment, is cleaved into latanoprost acid, a prostaglandin F2α receptor agonist, and butanediol mononitrate, a nitric oxide (NO)-donating moiety. LBN exhibited potent and effective IOP-lowering activity in three ocular hypertensive glaucoma animal models.13
NO donors relax the trabecular meshwork (TM) and increase aqueous humour outflow.14–17 They activate the large conductance calcium-activated potassium channel, or BKCa ion channel, involved in reducing TM cell volume.17–19 NO donors may trigger, among other things, reduction of actomyosin contractility and disassembly of the actin cytoskeleton and cell adhesion system in the cells of the conventional outflow pathway, causing cell shape changes and overall relaxation of the TM and inner wall of Schlemm’s canal leading to decreased resistance to aqueous humour outflow.8 ,20 ,21 Experimental data to date on the role of NO in modulating IOP through the conventional pathway were the focus of a recent review.17 In contrast, latanoprost acts by increasing the outflow of aqueous humour primarily through the uveoscleral pathway, consequently decreasing the IOP.22 There is a substantial reduction in IOP beginning approximately 3–4 h after latanoprost exposure and the reduction is sustained for at least 24 h.23
It is hypothesised that the use of the novel single entity, LBN, with the combined actions of latanoprost acid and NO will provide greater IOP reduction than latanoprost 0.005% while maintaining the convenience of a once daily dosing regimen. The objectives of the VOYAGER study were to evaluate the effectiveness and tolerability of varying concentrations of LBN compared with latanoprost 0.005%, and to determine the optimal concentration of LBN for IOP lowering as well as ocular and systemic safety in subjects with open angle glaucoma (OAG) or ocular hypertension (OHT).
Methods
Study design
The VOYAGER study (ClinicalTrials.gov identifier: NCT01223378) was a phase II, randomised, investigator-masked, parallel-group, dose-ranging study designed to compare the efficacy and safety of four different concentrations of LBN ophthalmic solution with Xalatan (latanoprost) 0.005% ophthalmic solution (Pfizer, New York, New York, USA) in subjects with OAG or OHT. The study was conducted across 23 investigative sites (15 in the USA and eight in the European Union). All sites received approval from their respective ethics committee (EC)/institutional review board (IRB). If a US study site was not associated with an EC/IRB, ethical approval was obtained from Schulman Associates IRB (Cincinnati, Ohio, USA). The clinical trial was conducted in accordance with Good Clinical Practice (as described by the International Conference on Harmonisation), the Code of Federal Regulations, the Declaration of Helsinki and applicable local regulations. All subjects provided written informed consent.
The study enrolled both currently treated and treatment-naive subjects (aged ≥18 years) diagnosed with OAG (including pigmentary or pseudoexfoliative) or OHT in one or both eyes. Subjects were eligible if they had an IOP of 22–32 mm Hg, and an IOP of ≥24 mm Hg for at least two of three measurements during Visit 3 (Day 1, baseline), which occurred after a 28-day washout period in subjects previously treated with IOP-lowering medications. Subjects had a best-corrected visual acuity (BCVA) of +0.7 logMAR (Snellen equivalent ∼20/100) or better in either eye.
Subjects were excluded if they had participated in any clinical trial within 30 days prior to screening and/or during the period of study participation; had a known hypersensitivity or contraindications to latanoprost or any of the ingredients in the study drugs or known contraindications to NO-donating treatment (ie, history of severe hypotension, alcohol abuse or hypersensitivity to any of the drug components); were unable to discontinue contact lens use during and for 15 min following instillation of study medication and during study visits; had central corneal thickness >600 μm in either eye or any condition that prevented reliable applanation tonometry (eg, significant corneal surface abnormalities) in either eye, advanced glaucoma (cup to disk ratio >0.8 or split fixation) or other significant ophthalmic disease. Subjects who required treatment with ocular or systemic corticosteroids or any other topical or systemic treatment for OAG or OHT, or had an anticipated need to initiate or modify medication that was known to affect IOP (eg, β-adrenergic antagonists, α-adrenergic agonists, calcium channel blockers, ACE inhibitors and angiotensin II receptor blockers) were also excluded.
Study treatments and assessments
Baseline data including demographics, relevant medical and ocular history, and concomitant medications were noted at Visit 1 (Screening). Eligible subjects being treated with an IOP-lowering medication were required to discontinue the agent for a minimum of 28 days prior to Visit 3 (Day 1/baseline) and participate in a mid-washout visit (Visit 2, Day −14). Pretreated subjects were withdrawn from the study if the mean or median IOP was >36 mm Hg in either eye at any point during the washout period. Eligible subjects were randomised into one of five treatment groups, including four different concentrations of LBN ophthalmic solution (0.006%, 0.012%, 0.024% and 0.040%) and latanoprost 0.005% ophthalmic solution. Subjects instilled one drop of study drug topically in the conjunctival sac(s) of the affected eye(s) once daily in the evening at approximately 20:00 for 28 days. If both eyes of a subject were eligible at baseline, both eyes were treated for the duration of study. The eye with the higher IOP at baseline was the study eye. When IOP was the same in both eyes, the right eye was the study eye.
Investigators were masked to the study medication. Because the active control bottle (Xalatan) was visibly different than the investigational bottles, a designee at each study site, other than the investigator, was responsible for the dispensing study treatment at Visit 3, instructing patients on proper instillation of study medication, and retrieval of materials at the end of the study. Attempts were made to mask the subjects by removing commercial labelling, replacing with identical investigational labels and packaging in identical kit boxes.
Upon randomisation, subjects completed five study visits: Visit 3 (Day 1/baseline), Visit 4 (Day 7±1 day), Visit 5 (Day 14±1 day), Visit 6 (Day 28±1 day) and Visit 7 (Day 29±1 day). At each visit, IOP was measured at 08:00, 12:00 and 16:00 using a Goldmann applanation tonometer, calibrated in accordance with the investigators’ standard of practice. Where possible, the same operator measured IOP, and the same tonometer was used at each visit for a given subject.
Safety assessments included adverse events (AEs), BCVA, ocular tolerability (ocular discomfort, burning sensation, pain, tearing and blurring of vision graded on a 6-point scale from 0 (none) to 5 (very severe)), ocular signs (biomicroscopy and ophthalmoscopy) and vital signs (blood pressure and heart rate). Treatment-emergent adverse events (TEAEs) were defined as AEs occurring on or after the first treatment dose. Vital signs and BCVA were assessed at each visit; ocular tolerability was assessed at dispensing and each follow-up visit. Slit lamp assessment of conjunctival hyperaemia (graded with the use of Institute for Eye Research photographic standards) and anterior chamber inflammation was performed prior to IOP measurement at each visit. Ophthalmoscopy was performed at screening randomisation and Day 28.
Endpoints
The primary efficacy endpoint was the reduction, or change from baseline (CFB), in mean diurnal IOP at Visit 6 (Day 28). Mean diurnal IOP was defined as the average of IOP at 08:00, 12:00 and 16:00. Secondary efficacy endpoints included reductions in mean diurnal IOP at Days 7, 14 and 29; reductions in IOP at specified time points (08:00, 12:00, 16:00); and the proportion of subjects with an IOP ≤18 mm Hg at all measurement time points.
The primary safety endpoint was the incidence of ocular and systemic AEs and their severity and relationship to the study drug.
Statistical analysis
Based on a prior phase II dose-finding study (ClinTrials.gov identifier: NCT00441883), a total of 350 subjects randomised into five treatment groups (70 subjects per arm) was calculated as providing adequate power (71%) to detect an IOP difference of 1.5 mm Hg, assuming an SD of 3.5 and significance level of 5%. To account for potential dropouts, 400 subjects (80 subjects per arm) were planned for enrolment to ensure at least 350 subjects completing the study.
The primary endpoint was evaluated for the intent-to-treat population, which comprised all randomised subjects, using an analysis of covariance with fixed-effect terms for baseline IOP and treatment. Each LBN treatment group was compared with the latanoprost group by computing a 95% CI around the differences between the least squares (LS) mean of each LBN treatment group and the LS mean of the latanoprost group. The difference was calculated as LBN minus the comparator latanoprost. The corresponding p values for each comparison are presented. The mean CFB in IOP for all treatment groups was compared with zero using paired two-sided t tests. No adjustments were made for multiple comparisons.
Safety analyses were based on the safety population, which comprised all randomised subjects who received ≥1 dose of the study drug. Ocular and non-ocular (systemic) TEAEs were described using discrete summaries at the eye and subject level, respectively, using the Medical Dictionary for Regulatory Activities (MedDRA) coding of the system organ class and preferred term for each treatment group as well as severity and relationship to study drug. Ocular signs and symptoms were summarised using discrete summary statistics by treatment groups. All statistical analyses were conducted using SAS software (SAS Institute, Cary, North Carolina, USA) V.9.1 or higher.
Results
Enrolment began in December 2010 and the last patient exited in December 2011. Of the 598 subjects screened, 413 subjects were randomised (n=82, LBN 0.006%; n=85, LBN 0.012%; n=83, LBN 0.024%; n=81, LBN 0.040%; n=82, latanoprost) and 396 subjects completed the study (n=76, LBN 0.006%; n=81, LBN 0.012%; n=80, LBN 0.024%; n=80, LBN 0.040%; n=79, latanoprost). The treatment groups were comparable with respect to demographics and baseline characteristics (table 1). The mean (SD) age of the subjects was 61.0 (11.44) years, with a higher percentage of female subjects. Subjects were predominantly non-Hispanic, non-Latino and Caucasian.
Subject demographics and baseline characteristics (intent-to-treat population)
A history of OHT or OAG/glaucoma was reported for 27.6% and 72.4% of study eyes, and 28.3% and 71.7% of fellow eyes, respectively. One subject had a history of iridotomy and approximately half (56.4%) of subjects were being treated with IOP-lowering medications at screening, most often prostaglandin analogues. We did not characterise these subjects in terms of their response to previous IOP-lowering medication. A total of 17.7% of subjects were being treated with latanoprost at screening and randomised across the five treatment groups. The majority (78.5%) of subjects were using systemic medications. Use of such medications was not altered during the trial and was comparable between the five groups.
Of the 17 randomised subjects who did not complete the study, six subjects discontinued due to AEs (n=3, LBN 0.006%; n=1, LBN 0.012%; n=1, LBN 0.024%; n=1, latanoprost), three subjects withdrew consent (n=1 each LBN 0.006%, LBN 0.040%, latanoprost), three subjects discontinued due to failure to follow the required study procedures (n=1 each LBN 0.006%, LBN 0.012%, LBN 0.024%), one subject was lost to follow-up (LBN 0.012%) and four subjects discontinued due to other reasons (n=1 each LBN 0.006%, LBN 0.012%, LBN 0.024%, latanoprost).
Efficacy
All treatments led to significant reductions in mean diurnal IOP from baseline at all follow-up visits (p<0.0001 paired t test). Among LBN groups, the IOP reductions were dose-dependent and appeared to plateau with the 0.024%–0.040% dose.
Table 2 presents the CFB in mean diurnal IOP at follow-up visits as well as the CFB at 08:00, 12:00 and 16:00 at these visits in study eyes. The LBN 0.024% and 0.040% treatment groups demonstrated a significantly greater reduction from baseline in diurnal IOP compared with the latanoprost group (p=0.005 and p=0.009, respectively) at Day 28, the primary endpoint (figure 1). While the LBN 0.006% and 0.012% groups also showed numerically greater reductions in IOP compared with the latanoprost group, these differences were not significant.
Reduction (mm Hg) from baseline in diurnal IOP and at 08:00, 12:00 and 16:00, Visits 4–7

Mean diurnal intraocular pressure (IOP) in the study eye at baseline and on Day 28 (intent-to-treat population). *p=0.005 versus latanoprost; †p=0.009 versus latanoprost. LBN, latanoprostene bunod.
Secondary efficacy evaluations demonstrated significantly greater reductions in mean diurnal IOP compared with latanoprost at Day 7 for the LBN 0.024% and 0.040% groups (p=0.033 and p=0.009, respectively), and at Day 14 for the LBN 0.024% group (p=0.015). Of note, at Day 29, 36–44 h after the last instillation of study medication, a marginally greater reduction in diurnal IOP was observed for the LBN 0.024% group (p=0.051).
Evaluations of reductions in IOP at specified time points showed significantly greater reductions in mean IOP compared with the latanoprost group for: the LBN 0.024% and 0.040% groups at 12:00 (p=0.032 and p=0.026, respectively) and 16:00 (p=0.017 and p=0.001, respectively) on Day 7; the LBN 0.024% group at 08:00 (p=0.016), 12:00 (p=0.028) and both the LBN 0.024% and 0.040% groups at 16:00 (p=0.027 and p=0.034, respectively) on Day 14; the LBN 0.040% group at 08:00 (p=0.049) and both the LBN 0.024% and 0.040% groups at 12:00 (p=0.006 and p=0.028, respectively) and 16:00 (p<0.001, p=0.002, respectively) on Day 28; and the LBN 0.024% group at 16:00 (p=0.045) on Day 29.
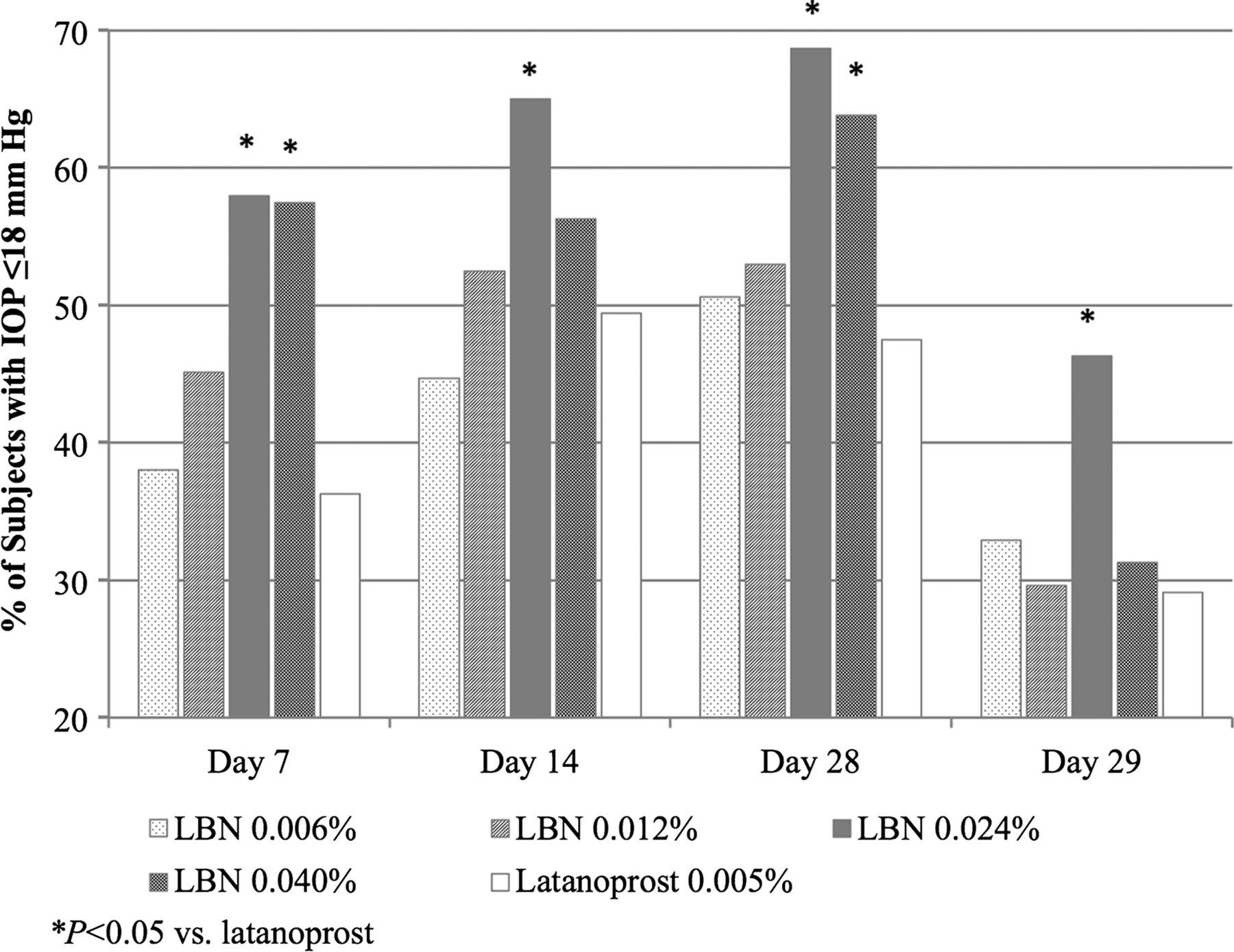
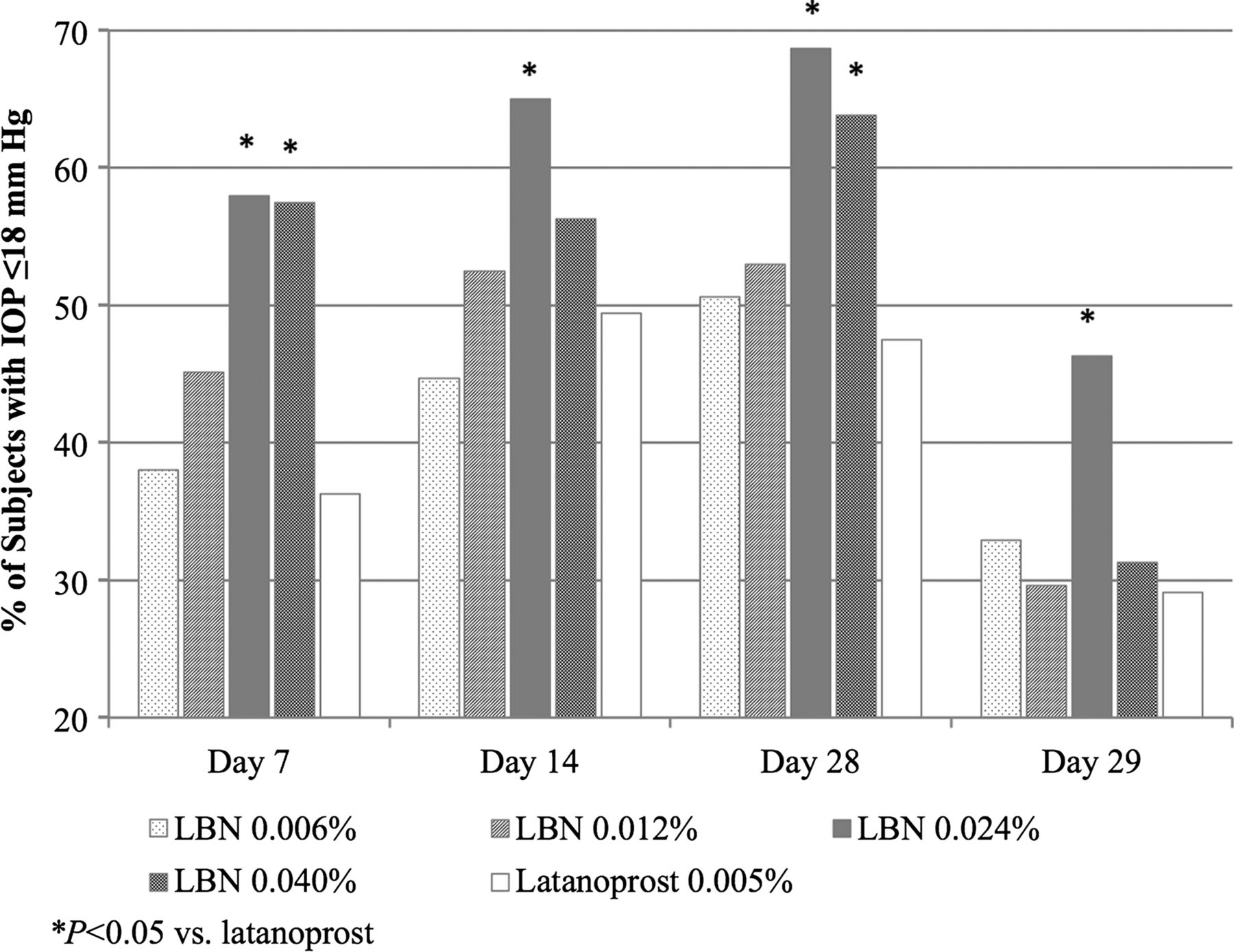
Figure 2 presents the proportion of subjects with a mean diurnal IOP ≤18 mm at each follow-up visit. A significantly greater proportion of subjects had mean diurnal IOP ≤18 mm Hg in the LBN 0.024% group at all visits (p≤0.046) and in the LBN 0.040% group at Day 7 (p=0.007) and Day 28 (p=0.039) compared with the latanoprost group.
{kind=link}
{kind=link}
Proportion of subjects with intraocular pressure (IOP) ≤18 mm Hg at follow-up visits (intent-to-treat population). *p<0.05 versus latanoprost. LBN, latanoprostene bunod.
Safety
Study drug exposure was similar across study groups whether evaluating days of exposure (range 26.9–27.5 days) or number of doses instilled (range 26.5–27.3). Nearly all subjects were fully compliant.
Overall, for the study eye, there was a numerically higher incidence of at least one ocular TEAE reported in each of the LBN treatment groups as compared with the latanoprost group (table 3). The most commonly reported ocular TEAE was instillation site pain (ie, drop instillation pain) reported by 14.6%, 16.7%, 12.0% and 17.3% of subjects in the LBN 0.006%, 0.012%, 0.024% and 0.040% groups, respectively, versus 6.1% of subjects in the latanoprost group. The most commonly reported ocular TEAE in the latanoprost group was ocular hyperaemia, reported for 8.5% of subjects versus 1.2%, 6.0%, 2.4% and 4.9% of subjects in the LBN 0.006%, 0.012%, 0.024% and 0.040% groups, respectively. Conjunctival hyperaemia was reported for 1.2%, 3.6%, 4.8% and 3.7% of subjects in the LBN 0.006%, 0.012%, 0.024% and 0.040% groups, respectively. Nearly all reports of instillation site pain and hyperaemia were considered drug related. Ocular TEAEs in treated fellow eyes were similar to those in study eyes. All ocular TEAEs were mild or moderate in severity.
Ocular TEAEs occurring in ≥2% of subjects in any treatment group
Non-ocular TEAEs were less commonly reported, and most were considered unrelated to study drug. The only non-ocular TEAE reported more than once and considered potentially related to study drug was headache (n=1 each, LBN 0.012%, LBN 0.024, latanoprost). Four serious non-ocular TEAEs were reported: gastric ulcer haemorrhage and acute myocardial infarction reported by one subject each in the latanoprost group and gastric ulcer and gastrointestinal haemorrhage reported by one subject in the LBN 0.006% group. All serious non-ocular TEAEs were considered unrelated/unlikely related to the study drug.
Six subjects discontinued from the study due to a TEAE. These included asthma (n=1, LBN 0.006%), migraine with aura (n=1, LBN 0.006%), instillation site pain (n=1, LBN 0.006%, n=1, LBN 0.012%), instillation site erythema (n=1, LBN 0.024%) and myocardial infarction (n=1, latanoprost).
Additional safety assessments (ie, ocular signs, ocular tolerability, BCVA and vital sign measurements) were unremarkable. Mean conjunctival hyperaemia severity, evaluated on a 4-point scale (none to severe), increased +0.1 to +0.3 from baseline across treatment groups, with no treatment differences in the proportion of subjects with hyperaemia.
Discussion
In the current study, LBN was efficacious at multiple concentrations, with dose-dependent IOP reductions over 28 days reaching a maximum effect with the 0.024% and 0.040% doses. For the primary endpoint—reduction in mean diurnal IOP at Day 28—statistically significant greater reductions were observed with LBN 0.024% and 0.040% compared with latanoprost 0.005%. Additionally, statistically significant differences in IOP reduction were noted between these LBN dose groups and latanoprost 0.005% for a majority of the secondary endpoints including CFB in mean diurnal IOP at earlier visits and CFB at specific time points during each visit, as well as for the proportion of subjects with IOP ≤18 mm Hg at follow-up visits. Both LBN 0.024% and 0.040% had comparable findings for the primary and secondary endpoints in this dose-ranging study. As the LBN 0.024% dose was the lower of the two most effective concentrations evaluated with greater IOP lowering and comparable side effects relative to latanoprost 0.005% solution, the LBN 0.024% dose was selected for further clinical evaluation in glaucoma and ocular hypertension.
In their study evaluating increasing concentrations of latanoprost in patients with OAG and OHT, Eveleth et al reported that increasing latanoprost concentrations to as high as 0.0125%, did not provide additional IOP lowering compared with latanoprost 0.005% and suggested that receptor saturation contributed to the lack of increased effect beyond the 0.005% dose.24 In the current study, there was no difference in treatment effect between LBN 0.006% and latanoprost 0.005%; these doses are equivalent in terms of molar concentrations (ie, 116–118 µM) and are therefore expected to deliver the same amount of latanoprost acid. That there was a dose-dependent increase in treatment effect with LBN beyond the 0.006% dose and up to the 0.024% dose suggests the dose–response curve for the NO-donating moiety of LBN, or butanediol mononitrate, includes a higher molar concentration range than that for latanoprost acid. It follows that improvements in IOP reduction observed with LBN 0.024% compared with latanoprost 0.005% likely reflect the additional action(s) of the NO-donating moiety. Further work is needed to clarify the extent of the contribution and precise mechanism of the additional IOP lowering due to butanediol mononitrate. In this regard, it is noteworthy that neither pupil dilation nor blurred vision was reported as AEs, suggesting that butanediol mononitrate did not alter IOP through relaxation of the ciliary muscle, and supporting a primary, direct effect of the NO-donating moiety on the TM/Schlemm's canal.
The Early Manifest Glaucoma Trial established that glaucoma progression was closely linked to the magnitude of the initial IOP reduction with treatment: each millimetre of mercury of IOP reduction from baseline in the first 3 months of treatment was associated with an approximate 10% decrease in visual field loss progression over the 6-year follow-up period.6 In our study, the difference in reduction of diurnal IOP between LBN 0.024% and latanoprost 0.005% was 1.23 mm Hg suggesting that treatment with LBN 0.024% is expected to have a greater effect on glaucoma progression than latanoprost.
The safety assessment indicated that LBN at concentrations from 0.006% to 0.040% dosed once daily for 28 days was well tolerated, although associated with slightly more TEAEs overall in the 0.040% treatment group. Hyperaemia, a common side effect of glaucoma hypotensive treatment, did not differ across treatments whether evaluated as a TEAE or by biomicroscopy. Instillation site pain, occurring more frequently with LBN treatments, did not affect compliance.
In conclusion, LBN 0.024% dosed once daily was the lower of the two most effective LBN doses evaluated with significantly greater IOP lowering compared with latanoprost 0.005% solution. To the best of our knowledge, this is the first phase II study that demonstrates a drug that is more effective for IOP lowering, without increased ocular hyperaemia and with comparable overall side effects, than the commercially available latanoprost 0.005% solution.
Acknowledgments
The authors thank the investigators who contributed to the acquisition of data and who along with the authors comprised the Voyager Study Group.
References
Footnotes
Collaborators Voyager Study Group: S Ackerman (Philadelphia, Pennsylvania, USA), J Branch (Winston-Salem, North Carolina, USA), A Cottingham (San Antonio, Texas, USA), D Day (Roswell, Georgia, USA), M Depenbusch (Pheonix, Arizona, USA), S El-Hazari (Glendale, California, USA), A Firozvi (Durham, North Carolina, USA), P Jorizzo (Medford, Oregon, USA), R Ou (Houston, Texas, USA), J Peace (Inglewood, California, USA), M Rotberg (Charlotte, North Carolina, USA), H Schenker (Rochester, New York, USA), S Smith (Fort Myers, Florida, USA), F Tyson (Cape Coral, Florida, USA), F Zaman (Houston, Texas, USA), L Madzharova (Sofia, Bulgaria), R Toshev (Varna, Bulgaria), P Vassilveva (Sofia, Bulgaria), M Misiuk-Hojło (Wrocław, Poland), J Kocięcki (Poznań, Poland), I Liehneova (Ústí nad Labem, Czech Republic) and E Růžičková (Praha, Czech Republic).
Contributors All authors contributed to the conception and design of the study, and to the analysis and interpretation of data. The authors retained full control of manuscript content, participated in drafting the manuscript and revising it for accuracy, and approved the final version submitted.
Funding This study was sponsored by Bausch + Lomb. Medical writing services, provided by Cactus Communications, were funded by Bausch + Lomb. Editorial assistance and data verification were provided by Mauricio Muñoz, PharmD and Linda Wang, PharmD.
Competing interests MM was a paid employee of Bausch + Lomb, Irvine, California, USA, during the conduct of this study. LW was a University of Southern California, School of Pharmacy fellow training at Bausch + Lomb during the preparation of this manuscript. RW received consulting fee from Aerie, Bausch + Lomb, Alcon, and Allergan, received grants from Genentech, Heidelberg Engineering, Quark, Nidek, National Eye Institute and Topcon. BS and TO are employees and hold stock at Bausch + Lomb. JV is an employee at Bausch + Lomb. KS received consulting fee from Allergan, Alcon, Bausch + Lomb and Santen. PK received consulting fee from Bausch + Lomb, Amakem, ONO Pharmaceuticals, Sucampo Pharmaceuticals, Sanofi-Fovea, Merck, Santen Pharmaceuticals, Valeant Pharma, Altheos and Kowa.
Ethics approval Schulman Associates IRB.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- At a glance