Article Text

Abstract
Aims: To determine the prevalence of cystoid macular oedema (CMO) in retinitis pigmentosa (RP) patients of various genetic subtypes using optical coherence tomography (OCT).
Methods: We performed a complete ocular examination on 124 RP patients including best corrected visual acuity, intraocular pressure measurement, anterior segment and a detailed fundus exam. OCT images were then acquired using two different units. The presence of hypo-reflective lacunae was used to diagnose CMO.
Results: Of the 124 patients, 47 showed CMO in at least one eye (38%), while 34 showed CMO in both eyes (27%). The prevalence of CMO in at least one eye for autosomal dominant (AD) patients was 52%, for autosomal recessive (AR) 39%, isolated 39%, Usher II 35% and none in the X linked recessive (XL) group. Using a chi-square analysis, no statistical significant difference was found for the prevalence of “bilateral CMO” (p = 0.60) or “CMO in at least one eye” (p = 0.59) among the AD, AR, isolated and Usher II genetic subtypes.
Conclusion: Because of its notable prevalence, it would seem prudent to screen RP patients by OCT for the possible presence of CMO, to identify those amenable to treatment and also for future treatment trials when monitoring visual acuity.
Statistics from Altmetric.com
Retinitis pigmentosa (RP) is a group of hereditary progressive retinal diseases caused by gene abnormalities on several different chromosomes resulting in the progressive loss of photoreceptors, which leads to impaired night vision and a gradual loss of visual field.1–4 In addition to attenuated retinal vessels, atrophic changes in optic disc and bone spicule-like pigment clumping, macular cysts have been reported to be associated with RP.5 6
The prevalence of cystoid macular oedema (CMO) in RP patients has most commonly been reported to be from 11% to 20% using fluorescein angiography (FA) and fundus exam.2 6 Hirakawa et al7 reported the prevalence of CMO in RP patients using optical coherence tomography (OCT) to be 13%. However, in their study, only horizontal and vertical scanning lines were acquired through the fovea, which could have underestimated the presence of juxtafoveal cystic lesions in the other meridians. Adackapara et al8 reported a 49% prevalence of CMO using OCT in 39 RP patients with a visual acuity better than 20/100. Theses authors might have overestimated the presence of CMO because the presence of a single central cyst on one OCT evaluation was considered as CMO.
OCT provides an objective cross-sectional examination of the fundus,9 which can serve as a useful tool for objectively monitoring macular oedema.10 With the advent of Time Domain, and more recently Fourier Domain OCT technology, there is an opportunity for a more comprehensive and non-invasive way to evaluate the presence of macular oedema without the routine use of FA.9 11–13
Therefore, in the current study we sought to determine the prevalence of cystoid macular oedema in RP patients of various genetic subtypes using OCT technology.
MATERIALS AND METHODS
The total number of subjects ultimately included in this study was 124 RP patients from ages 8 to 71 years. This included all RP patients who were seen by the authors between 20 June 2007 and 8 January 2008. Out of these patients, 18 were new patients who were just diagnosed as having RP, and 84 were former patients who returned for their general routine visit and were asked to participate in this study. Out of these 84 RP patients, 12 were previously diagnosed as having macular oedema, and they were on treatment for CMO. These patients were included in our study when they returned to our clinic as a part of their routine follow-up visit to monitor any cystic changes.
We additionally reviewed the charts and attempted to contact all RP patients seen by one of the authors (GAF) within the previous 5 years and who had neither cystic nor atrophic-appearing macular changes on prior funduscopic exams. Out of 98 charts that were reviewed, we were able to contact and re-evaluate prospectively 22 patients who were asked to return for a re-examination of their macula and OCT testing for the presence of CMO.
The diagnosis of RP was based on the patients’ history of poor night vision, side vision restriction, markedly reduced or non-deductible a- and b-wave amplitudes on ERG testing, in addition to ophthalmoscopic findings including characteristic fundus changes of attenuated retinal vessels and bone spicule-like pigment clumping. We excluded any patient with uveitis, diabetic retinopathy or any disease that could cause RP-like fundus changes. We also excluded all aphakic and pseudophakic patients, and all syndromic forms of RP except for type II Usher syndrome. Thirty-two additional patients who came for their routine annual visit were not included in this study due to our inability to obtain OCT images on either eye as a consequence of either dense cataracts, a lack of proper fixation or markedly reduced visual acuity. One eye only on two of the patients was included due to an inability to obtain images on the other eye.
The 124 RP patients were categorised into various genetic subtypes as described in previous reports.14 15 Included were 31 patients from 27 families with autosomal dominant (AD) inheritance, 18 patients with autosomal recessive (AR) inheritance, nine patients with X linked recessive inheritance (XL), and 39 patients from 39 different families who showed no evidence of other affected family members and therefore were classified as an isolated subtype. We also included 23 patients from 22 families with type II Usher syndrome. The inheritance pattern for four of the included patients could not be determined. None of the patients in the AR subgroup showed a phenotype consistent with enhanced S-cone syndrome or other progressive retinal degenerations.
Informed consent was obtained from all subjects after an explanation of the nature and possible consequences of the study. All patients then had a complete eye exam including best corrected visual acuity using an Early Treatment Diabetic Retinopathy Study chart (ETDRS). Anterior segment exam, intraocular pressure measurement using Goldmann applanation tonometry, and a dilated fundus exam by direct ophthalmoscopy, binocular indirect and 78 dioptre non-contact lens were performed. OCT images were then acquired by experienced examiners using one of two different OCT instruments. On 112 of the 124 patients, Fourier Domain OCT (FD-OCT) was performed using an RTVue model RT100 (Optovue, Fremont, CA; software version 1.2.6 and 2.0.3.2). This instrument provides a high-speed acquisition time of 26 000 A-scans per second and a high-depth-resolution retinal scanner (5 µm). The protocol used on FD-OCT was the radial slicer that acquires simultaneously (within 0.27 s) twelve 6 mm radial line scans through the centre of the fovea at 15° intervals using internal fixation, where every radial scan consists of an average of 1024 A-scans. Only images with good quality (signal strength index >40) were included in this study. Twelve additional patients were scanned using a Time-Domain (TD) OCT, OCT3 commercial instrument (StratusOCT, software version 4.0.1; Carl Zeiss Meditec, Dublin, CA) using the macular thickness protocol where six 6 mm radial scans were acquired on each eye at 30° intervals centred on the patient’s fovea. All the accepted scans had signal intensity strengths of five or more. A determination for the presence of CMO on OCT scans, using either instrument, included the presence of several cystoid spaces as hypo-reflective lacunae with well-defined boundaries on at least two radial scans in the macular area. The foveal thickness for those who had CMO in both eyes was averaged for each patient, while in those who had unilateral CMO only the foveal thickness in that eye was recorded for analysis using one-way ANOVA.
This project was conducted in the Department of Ophthalmology at the University of Illinois at Chicago; it was approved by an institutional review board and the University of Illinois ethics committee, and was performed in accordance with the tenets of the Declaration of Helsinki.
RESULTS
Included were 59 males (48%) and 65 females (52%). This study group was comprised of 95 Caucasian (77%), 18 African–American (15%), eight Hispanic (6%), and three Asian patients (2%). The average best-corrected visual acuity for all patients was 0.48 logMAR units, equivalent to 20/60 on a Snellen acuity chart (range, −0.04 to 2.7, equivalent to from 20/20[+2] to light perception on a Snellen chart).
Out of the 124 RP patients included, 47 showed CMO on OCT testing in at least one eye (38%), and 34 showed CMO in both eyes (27%). Within the genetic groups, 12 out of the 31 AD (age range 12–71 years) had bilateral CMO (39%) and 16 patients had CMO in at least one eye (52%). In the AR group (age range 12–67 years), four out of the 18 patients had bilateral CMO (22%) and seven patients had CMO in at least one eye (39%). In the isolated group (age range 8–67 years), 11 out of the 39 patients had bilateral CMO (28%) and 15 patients had CMO in at least one eye (39%). In the Usher II group (age range 9–53 years), six out of the 23 patients had bilateral CMO (26%) and eight patients had CMO in at least one eye (35%). None of the patients in the XL group (age range 9–65 years) showed CMO in either eye, and one of the four patients with undetermined genetic type (age range 14–52 years) had CMO in both eyes (25%). The modes of inheritance, the number and percentage of patients with CMO, and the age range of the patients in each genetic group are summarised in table 1.
The number of families included matched the number of patients except for the AD and the Usher II groups. In the AD group, 10 out of the 27 families had at least one member with bilateral CMO (37%), and 14 families had at least one member with CMO in at least one eye (52%). In the Usher II group, six out of the 22 families had at least one member with bilateral CMO (27%), and eight families had at least one member with CMO in at least one eye (36%).
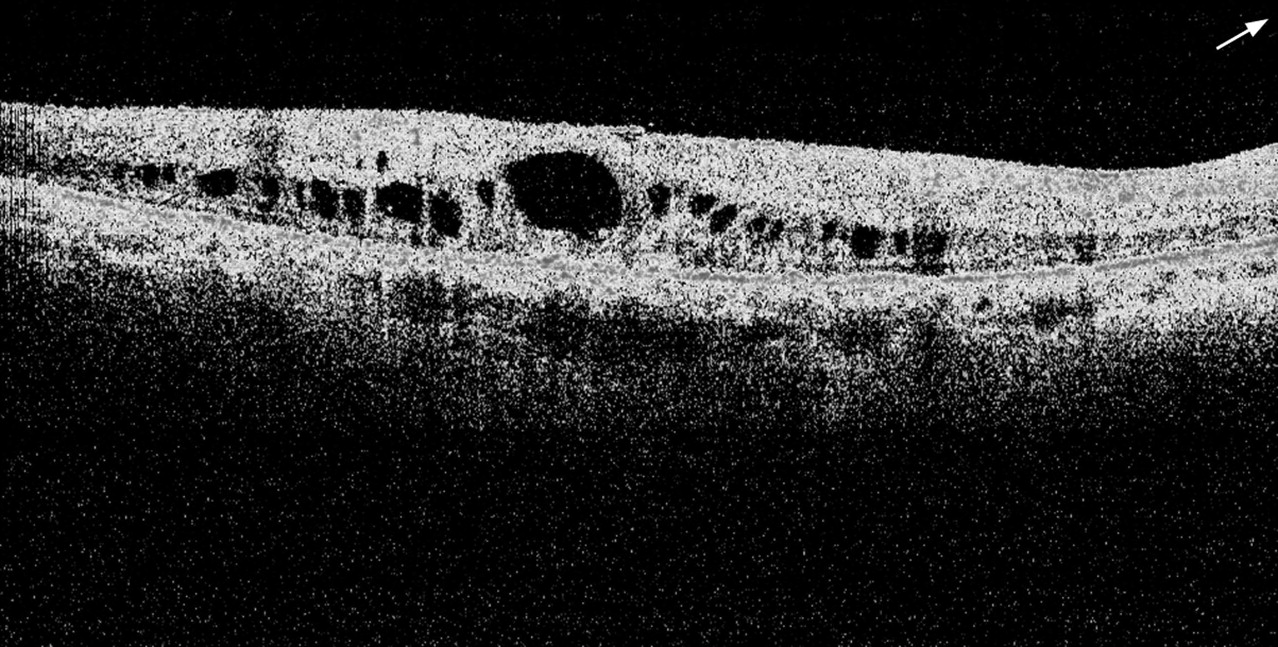
The average age of the patients included in this study was 38 years. There were 10 patients within the age group 1 to 14 years (8.1%), four of them had bilateral CMO (40%), and six had CMO in at least one eye (60%); 31 patients were within the age group 15 to 29 years (25%), of whom seven had bilateral CMO (23%), and 10 had CMO in at least one eye (32%); there were 34 patients within the age group 30 to 44 (27.4%), out of whom eight patients had bilateral CMO (24%), and 10 had CMO in at least one eye (30%); in 40 patients within the age group 45 to 59 years (32.2%), there were 12 who had bilateral CMO (30%) and 17 had CMO in at least one eye (42%). The remaining nine patients (7.3%) of the 124 RP patients were 60 years or older, three had bilateral CMO (33%), and four had CMO in at least one eye (44%). Table 2 summarises the number and percentage of patients with CMO in each of the different age groups. Figure 1 displays an FD-OCT image with cystic-appearing spaces (foveal thickness 422 µm) in the right eye of one of the RP patients included in the isolated group.
{kind=link}
The average foveal thickness in patients who had CMO in at least one eye was 316 (SD 105) µm (range 167–685). Table 3 summarises the foveal thickness average, SD and range within each genetic subgroup (AD, AR, Isolated, and Usher II) in those who had CMO in at least one eye. No significant differences in foveal thickness were detected (p = 0.38) among those genetic groups.
Because of the relatively small number of patients included in the XL and the genetically unknown groups, we did not include those groups in the comparison of the prevalence of CMO among the different genetic subtypes. Using chi-square analysis, no statistical significance was obtained for the comparison of the rate of CMO among the AD, AR, isolated and Usher II genetic subtypes, using either the bilateral CMO criterion (p = 0.60) or the “at least one eye with CMO” criterion (p = 0.59).
DISCUSSION
OCT was previously reported to detect the presence of macular oedema, independent of the fluorescein angiographic degree of leakage,7 and observed to be at least as sensitive as fluorescein angiography for identifying macular oedema.11 16
Our study indicates that as many as 38% of RP patients may manifest cystic macular changes on OCT testing in at least one eye, while 27% of our RP cohort showed such changes in both eyes. This observed prevalence of CMO is more than what has been reported in the literature using FA alone,2 6 and also exceeds what was found using OCT horizontal and vertical scans only.7 This is more likely due to the sensitivity of the OCT in detecting cystic lesions in different meridians around the fovea, which cannot be observed clinically by funduscopic exam, and may not show well, or at all, on FA. Further, our prevalence for CMO is less than what was reported by Adackapara et al8 (49%), which could, in part, be attributed to our larger number of patients and our criteria for identifying CMO, where a single cyst on a single radial scan was not considered as CMO. Nevertheless, we may also have actually underestimated the presence of macular oedema in our cohort of patients because we did not include patients who had increased macular thickness with an absence of cystoid changes as having CMO, since increased macular thickness does not always indicate the presence of non-cystic macular oedema. Rather it could be a result of secondary glial cell proliferation or other anatomic alteration. There was no statistically significant difference in foveal thickness in those who had CMO in at least one eye among the different genetic subtypes (p = 0.38).
A previous study reported that macular oedema did not appear to be more common in any of the genetic subtypes of RP.17 In this study, we found the proportion of RP patients with CMO in at least one eye on OCT testing within each genetic group was maximal for the autosomal dominant group (52%), followed by the autosomal recessive (39%), isolated (39%) and Usher II (35%). However, no statistical significance was found when we compared the prevalence of CMO among the AD, AR, isolated and Usher II groups using the criterion of a bilateral CMO (p = 0.60), or even the criterion of CMO in at least one eye (p = 0.59). None of the nine patients in the X linked genetic subtype showed CMO changes, which is consistent with a previous report,2 even though this latter subtype is considered to show the most severe form of disease in terms of visual impairment.18 Sandberg and co-workers (ARVO Abstract 3723, 2007) did not observe macular cysts in patients with X linked disease, and they documented a 28% prevalence of CMO, in at least one eye, in RP patients using OCT. This compares with 38% observed in our cohort.
Although the number of patients in the youngest and the oldest age groups were small, the presence of CMO did not show a predominance within any age-group category. Nevertheless, it is conceivable that a different cohort of RP patients whose age range differs from those in our population might show a somewhat different percentage for the prevalence of CMO.
Because of the high prevalence of CMO found in our cohort of RP patients, it would seem prudent to screen such patients with OCT to define the possible presence of macular oedema so that treatment can be implemented in certain patients who may benefit from its use.19–24 Further, screening would also be useful for future treatment trials targeted to improve photoreceptor cell function.
REFERENCES
Footnotes
Funding: Supported by funds from the Foundation Fighting Blindness, Owings Mills, Maryland; Grant Healthcare Foundation, Lake Forest, Illinois; NIH core grant EYOO1792; and an unrestricted departmental grant from Research to Prevent Blindness. The funding source had no involvement in the design of this study; in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Competing interests: None.
Ethics approval: This study was approved by an institutional review board and the University of Illinois ethics committee, and was performed in accordance with the tenets of the Declaration of Helsinki.
Patient consent: Obtained.