Article Text

Abstract
Aims: To search for mutations in the frizzled 4 (FZD4) gene in patients with familial exudative vitreoretinopathy (FEVR) and to delineate the defective gene associated clinical features.
Methods: Direct sequencing following polymerase chain reaction of exons of FZD4 was performed for 24 probands with FEVR (18 familial and six sporadic), and some of their families. Clinical symptoms among individuals with mutations were assessed.
Results: Four novel mutations were identified in four patients with familial and one with sporadic FEVR. Three of these mutations were missense (M105V, R417Q, and G488D) and one was a nonsense change (W319X). M105V, R417Q, and G488D co-segregated with the disease. None of these sequence changes was found among 300 chromosomes from 150 healthy volunteers. The severity of vitreoretinopathy in the individuals involved in this study varied, but no patient with mutations in FZD4 exhibited rhegmatogenous retinal detachment although this pathology is thought to be the most common type of retinal detachment in FEVR.
Conclusion:FZD4 gene mutations were found in some cases of autosomal dominant and sporadic FEVR. FZD4 mutations were responsible for FEVR with variable clinical manifestations.
- gene mutations
- retinal detachment
- familial exudative vitreoretinopathy
Statistics from Altmetric.com
Familial exudative vitreoretinopathy (FEVR) is a hereditary vitreoretinal disorder, first described by Criswick and Schepens.1 The disease manifests as various retinal pathologies, such as retinal exudates, retinal neovascularisation, peripheral fibrovascular mass, macular ectopia, retinal fold, retinal detachment, and vitreous haemorrhage, in the first decade of life.2–4 FEVR may also include greater numbers of patients with rhegmatogenous retinal detachment in the second and third decades of life.3–5 The terminal stage of severely affected eyes is characterised by chronic retinal detachment, leading to total blindness. Although the penetrance of the disease is thought to be nearly 100%, the expressivity differs widely both between and within families.6 Most individuals remain completely asymptomatic and the only consistent sign is non-perfusion of the retinal periphery.7 Such a mild change is only accurately detectable by fluorescein angiography, and the difficulty in distinguishing affected individuals without vision loss from unaffected individuals can hamper the diagnosis of FEVR.6 The nature of this genetic disorder and the underlying pathophysiology are unknown. There is a striking phenotypic similarity between FEVR and the retinopathy of prematurity. However, patients with FEVR have no history of prematurity or supplemental oxygen therapy in the neonatal period.8
FEVR is genetically heterogeneous, as are other retinal dystrophies, such as retinitis pigmentosa.9 Autosomal dominant inheritance is regarded as the major heritability pattern for FEVR, but X linked recessive and autosomal recessive forms have also been reported, as well as sporadic cases.10–12 To date, at least three loci for the FEVR gene have been reported—that is, on chromosomes 11q13–23 (EVR1), Xp11.4 (EVR2), and 11p13–12 (EVR3).13–15 The EVR2 gene has been shown to be identical to the Norrie disease gene (NDP).16 Robitaille et al have recently identified the EVR1 gene as the frizzled 4 gene (FZD4), a human homologue of a cell surface receptor of Wnt signalling in Drosophila.17 So far, two independent mutations in the FZD4 gene have been identified in two families. However, the incidence of mutations in this gene among patients with FEVR is unclear. We therefore looked for mutations in the FZD4 gene in patients with the disease, and investigated the associated phenotypic expressivity.
SUBJECTS AND METHODS
Patients were considered to have FEVR if they exhibited at least one of the typical clinical findings: severe retinal exudate, retinal neovascularisation, peripheral fibrovascular mass, macular ectopia, retinal fold, retinal detachment, or vitreous haemorrhage. Other possible causes of peripheral retinal pathology, such as the retinopathy of prematurity, were excluded before diagnosis. All patients were Japanese. Twenty had familial FEVR (20 families), whereas six lacked any apparent family history of dominant inheritance and were therefore diagnosed as suffering sporadic FEVR. Before this study, some of the participants had been genotyped with 11q13–23 markers, and screened for mutations in the NDP gene.18 The disease in 11 out of 20 families was shown to be linked to 11q13–23 and that in two families was shown to be unlinked, and thus the subjects in the latter were excluded from the present study. No mutation in the NDP gene has yet to be found in this study. Therefore, a total of 24 patients participated in this study. All patients were examined at Fukuoka University Hospital. The study followed the tenets of the Declaration of Helsinki. Informed consent was obtained from all subjects involved in the study, and the protocol was approved by the ethics review board of Fukuoka University. Ocular examinations included refraction, visual acuity, intraocular pressure, slit lamp, fundus, and ultrasonographic examinations. Fluorescein angiography was performed on several individuals. DNA samples were extracted from peripheral blood using a DNA extraction kit (QiaAmp, Qiagen, Chatsworth, CA, USA) or the ethanol precipitation method.19 To identify mutations in the exons of the FZD4 gene, oligonucleotide primers based on the flanking intron/untranslated region (UTR) sequences were designed (see Table 1) with the Primer3 program (Whitehead Institute for Biomedical Research/MIT Center for Genome Research, Cambridge, MA, USA). Polymerase chain reaction (PCR) was performed essentially as previously described.20 An annealing temperature of 65°C was used for all fragments, and 5% dimethylsulfoxide was added for the amplification of the exon 1 fragment. Direct sequencing was performed using the BigDye Terminator Sequencing Kit (PE Biosystems, Foster City, CA) after treatment with shrimp alkaline phosphatase (Roche Diagnostics, Mannheim, Germany) and exonuclease I (New England Biolabs, Beverly, MA, USA). The samples were denatured and analysed with a DNA sequencer (3700 Genetic Analyser; PE Biosystems). Once a mutation was detected, samples from the rest of the family members were analysed.
Sequences of PCR primers used to amplify FZD4 coding segments
RESULTS
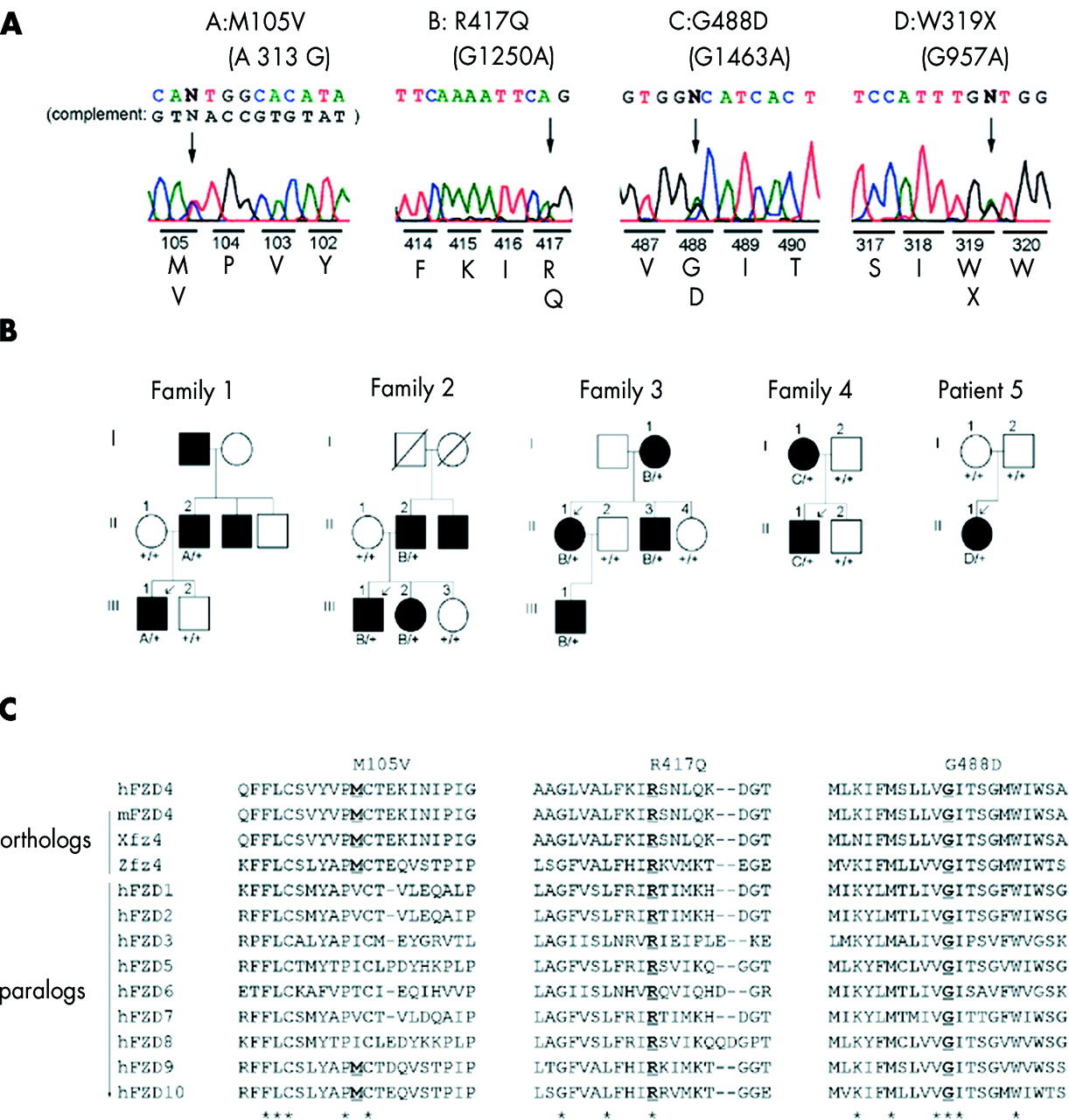
We surveyed 24 unrelated patients with FEVR for mutations in the FZD4 gene. Four novel mutations were found in five probands (four with familial and one with sporadic FEVR) in the coding sequence of the FZD4 gene (Figs 1 and 2). Three of these were missense changes: M105V (A313G), R417Q (G1250A), and G488D (G1463A); and one was a nonsense change, W319X (G957A). Further analysis of family members revealed a total of 12 mutations (Fig 1 and Table 2). All of the missense mutations co-segregated with the disease in families 1–4 (Fig 1A and 1B). W319X was found in the proband of family 5, and no sequence change was found in either parent, suggesting a new germinal mutation. The FZD4 protein sequences of humans and other species were aligned against sequences of other human FZD protein family members, using Clustal W software (Fig 1C).21 All mutated residues were located at residues conserved among humans and other species (mouse, zebra fish, and frog).17 All mutated residues, except codon 105, were also conserved among the 10 proteins of the human FZD family (FZD1–FZD10).17,22 None of the sequence changes were found in 300 chromosomes sampled from 150 healthy volunteers. The clinical symptoms of the 12 individuals with the 12 mutations in the FZD4 gene are summarised in Table 2. Of these 12 individuals, five (38%) were asymptomatic, and the severity of FEVR varied among the 24 eyes of the 12 individuals. Various forms of retinal detachment were seen in nine eyes: five eyes had falciform retinal folds, three eyes had chronic total retinal atrophy from infancy, and one eye had exudative retinal detachment. However, none exhibited rhegmatogenous retinal detachment.
Mutations in FZD4 gene and the associated clinical findings

(A) Novel mutations in the FZD4 gene in patients with FEVR. The altered amino acids with their positions and corresponding nucleotide changes are shown with the sequencing trace data. M105V is shown in the antisense direction (the complement nucleotides in the sense direction is also shown), and the other mutations are shown in sense direction. Arrows indicate the positions of altered nucleotides. (B) Schematic representations of the pedigrees of four families, illustrating the co-segregation of the FZD4 mutations (M105V, R417Q, G488D) with FEVR (families 1–4), as well as one sporadic case (patient 5). Solid symbols indicate individuals with a diagnosis of FEVR. Arrows indicate probands. Individuals from whom sequence data were obtained are numbered. Plus signs indicate the wild type allele, and A, B, C, and D indicate the sequence changes M105V, R417Q, G488D, and W319X, respectively, which are also indicated above the trace data at the top. (C) An amino acid sequence alignment of human, mouse, frog, and zebra fish of FZD4 as well as homologues from human FZD family in the vicinity of each missense mutation. The affected amino acid is underlined. Conserved amino acids are indicated by an asterisk below the alignment. Sequence data were derived from Genbank based on the reference by Robitaille et al.17

Schematic diagram of the locations of seven sequence changes identified in the FZD4 gene in FEVR patients. Four mutations (underlined) and one polymorphism (in italics) identified in this study are at the top. Mutations previously reported by Robitaille et al17 are at the bottom.
The proband of family 1 (M105V) was a 4 year old male who weighed 3232 g at birth after 39 weeks of gestation, with poor vision in both eyes since infancy. Bilateral vitreous opacity and retinal exudates were observed. Macular ectopia was visible in the right eye and a falciform retinal fold in the left eye. The father had peripheral avascularisation with a typical scalloped border in both eyes. Furthermore, the sibling of the father was amblyopic with a history of retinal detachment. The proband of family 2 (R417Q) was an 18 year old male who was born at a normal gestational age weighing 3000 g. Poor vision was present in both eyes from infancy. The right eye revealed a severe falciform retinal fold with a peripheral fibrovascular mass, and the best corrected visual acuity was 0.05 (Fig 3A). The left eye was totally blind, with posterior synechiae and chronic retinal detachment. The right eye of the father was blind as a result of glaucoma associated with chronic retinal detachment. His left eye showed peripheral avascularisation. One of the proband’s siblings was completely asymptomatic, with bilateral peripheral retinas similar to those in the father’s left eye. A brother of the father was totally blind in both eyes. The proband of family 3 (R417Q), a 21 year old female, had remarkable retinal exudate in the right eye and retinal holes in the peripheral avascular retina in both eyes (Fig 3B). The visual acuity in the right eye was reduced to light perception. All other affected family members had vitreous membranes and peripheral avascularisation in both eyes, with no symptoms, except for the mother who had mild vitreous opacity in the right eye. The proband of family 4 (G488D), a 1 year old male, had falciform retinal folds in both eyes (Fig 3C). The mother had a small retinal hole with mild avascularisation in the temporal peripheral retina of the right eye, without the typical scalloped border. The proband of family 5 (W319X), a 1 year old female, had a falciform retinal fold in the left eye and chronic retinal detachment in the right eye. The parents presented with normal retinal changes.

(A) Fundus image of the right eye of the proband of family 2 (III:1), showing a falciform retinal fold associated with a fibrovascular mass. (B) The right eye of the proband of family 3 (II:1), showing severe retinal exudate. (C) The left eye of the proband of family 4 (II:1), showing a falciform retinal fold.
In addition to the mutations described above, a missense change, H69Y (C205T) was found in the proband of family 4 (Fig 4). Thus, the patient possessed double sequence change—that is, G488D from the affected mother and H69Y from the unaffected father (Fig 4B). H69Y was also found in the patient with sporadic FEVR (patient 6), for which parent data were not available. This patient was a 38 year old female with bilateral macular ectopia and peripheral degeneration, with an avascular retina (Fig 4C). There was no family history of retinal detachment. Although the mutated residue, codon 69 was highly conserved as shown in Figure 4D H69Y was detected in two individuals out of 150 healthy volunteers whose fundi were not examined. We suspect this sequence change may have some causative effects as discussed later.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
A missense change (H69Y) detected in the FZD4 gene in family 4 and a patient with sporadic FEVR (patient 6). (A) An altered amino acid with the position from the proband of family 4. (B) Schematic pedigrees of family 4 and patient 6. C and E indicate G488D (referable to Fig 1) and H69Y, respectively. (C) The right eye of the patient, showing an ectopic macula. (D) An amino acid sequence alignment of human, mouse, frog, and zebra fish of FZD4 as well as homologues from human FZD family in the vicinity of codon 69. See Figure 1 for explanation.
DISCUSSION
We examined 24 patients with FEVR for mutations in the FZD4 gene. Four patients with familial FEVR and one with sporadic FEVR had a total of four novel mutations in the FZD4 gene, three of which were missense mutations and the other a nonsense mutation. All the corresponding amino acid residues were highly conserved, and all but codon 417 were located in functional domains—the cysteine rich domain (M105V) and transmembrane domains (W319X and G488D).17,22 These sequence changes were not found in 150 normal individuals (300 chromosomes), with which a polymorphism of 1% frequency would not be missed between 5% and 20% of the time.23 These findings suggest that the sequence changes found in this study may be pathogenic.
As shown in Table 2, the expressivity of FZD4 mutations differed widely both among and between families, and one third of the affected individuals were asymptomatic. Although rhegmatogenous retinal detachment is thought to be the most common type of retinal detachment in FEVR,3–5 none of the patients with FZD4 mutations in this study displayed rhegmatogenous retinal detachment. This finding is consistent with the results of the large Canadian family in which the FZD4 mutation was first identified.17,24 Therefore, the FZD4 mutation is responsible for FEVR with variable clinical manifestations, but does not explain notable instances of the disease, which involve rhegmatogenous retinal detachment.
Codon 69 of FZD4 was located in the cysteine rich domain and highly conserved. These findings suggest that the missense change, H69Y may be pathogenic. However, H69Y was also found in healthy individuals and might constitute a polymorphism. An alternative interpretation of H69Y is that it is pathogenic with low penetrance, because this change was found with higher frequency in patients with FEVR than in the normal population. Consistent with this assumption, the proband of family 4, to whom H69Y and a second change (G488D) were heterozygously transmitted, presented with a more severe phenotype than the mother who carried a single G488D mutation as a result of the compound heterozygosity. Further investigation is required to evaluate the functional significance of this sequence change.
Contrary to our previous study indicating that 11 out of 18 families with FEVR were linked to the EVR1 locus (Kondo et al18 and our unpublished data), we did not detect any sequence changes in the FZD4 exons in eight out of 11 families. In these eight families, the responsible mutations may occur in non-coding regions of the FZD4 gene that are involved in regulatory processes, or in other genes linked to FZD4. Further identification of the mutation spectrum of this FEVR causing gene and its associations with the clinical features described in this study should provide the information necessary for appropriate genetic counselling, as well as possible prophylactic treatments for retinal detachment in individuals with this genetic predisposition.
Acknowledgments
The authors thank the family members who participated in this study for their cooperation.