Article Text

Abstract
The last decade has been an age of enlightenment as far as mitochondrial pathology is concerned. Well established nuclear genetic diseases, such as Friedreich’s ataxia,1 2 Wilson disease,3 and autosomal recessive hereditary spastic paraplegia,4 have been shown to have a mitochondrial basis, and we are just starting to unravel the complex nuclear genetic disorders which directly cause mitochondrial dysfunction (table 1). However, in addition to the 3 billion base pair nuclear genome, each human cell typically contains thousands of copies of a small, 16.5 kb circular molecule of double stranded DNA (fig 1). Mitochondrial DNA (mtDNA) accounts for only 1% of the total cellular nucleic acid content. It encodes for 13 polypeptides which are essential for aerobic metabolism and defects of the mitochondrial genome are an important cause of human disease.92 93 Since the characterisation of the first pathogenic mtDNA defects in 1988,5 13 over 50 point mutations and well over 100 rearrangements of the mitochondrial genome have been associated with human disease94 95 (http://www.gen.emory.edu/mitomap.html). These disorders form the focus of this article.
- mitochondrial DNA
- mitochondrial disease
- heteroplasmy
- genetic counselling
Statistics from Altmetric.com
Many mtDNA diseases present with neurological symptoms and signs, but the clinical features are often non-specific and diffuse, and mitochondrial diseases have frequently been misdiagnosed when only clinical abnormalities are considered. MtDNA defects may present to physicians in any specialty and many of these disorders appear to be sporadic. It is precisely because of the diverse clinical features associated with pathogenic mtDNA defects that, not unreasonably, mitochondrial disease enters the differential diagnosis of so many conditions.96 This marked clinical heterogeneity creates difficulties for the clinician. Unlike nuclear genetic defects, which can often be diagnosed with a simple blood test, the investigation of mitochondrial disease is far more complex, and molecular genetic studies play only one part in the multidisciplinary approach that should be used for these patients.97
This articles focuses on three fundamental questions of clinical mitochondrial genetics. (1) When should we look for mitochondrial disease? (2) How should we investigate patients with suspected mitochondrial disease? (3) How should we manage patients with mtDNA disease?
The clinical features of mtDNA disease
Patients with mtDNA disease fall into three main categories. In the minority of cases, it is relatively straightforward to identify that a clinical syndrome involves mitochondrial dysfunction and these constitute the first category. Classically, patients with MELAS (MitochondrialEncephalomyopathy withLactic Acidosis and Stroke-like episodes) proceed through their normal motor and cognitive milestones but their stature usually remains short. They often develop bilateral deafness in their late childhood or teenage years, and they present with diabetes, seizures, stroke-like episodes, and an encephalopathy in their third or fourth decade.98 Not all MELAS patients, however, fit this pattern and diabetes and deafness may be the only features,99 and a cardiomyopathy may be missed.100 As another example, patients withLeber HereditaryOptic Neuropathy (LHON) are more commonly male, and they usually present in their second or third decade with acute or subacute bilateral visual failure, which may recover to some degree in the following months.101Although these presentations are “classical”, it cannot be stressed too heavily that mtDNA diseases are phenotypically diverse. Thus, the first feature of MELAS may be a stroke-like episode in late middle age,102 103 and LHON may present in the seventh decade or it may have an atypically insidious onset.104
There are other “classical” mitochondrial disorders. External ophthalmoplegia is a relatively common feature of patients with a mitochondrial encephalomyopathy.97 This abnormality may present in the second decade in association with ataxia, bilateral deafness, a cardiac conduction defect (fig 2), and high CSF protein (as in Kearns-Sayre syndrome, fig 3). More often, it occurs in isolation or with ptosis and a mild proximal myopathy (CPEO, orChronicProgressiveExternalOphthalmoplegia).6 Two other syndromes warrant a mention, a neuropathy with ataxia and a pigmentary retinopathy and ataxia is suggestive of NARP (Neurogenic weaknessAtaxia withRetinitisPigmentosa),23 and myoclonic epilepsy with ataxia and a myopathy are prominent features of MERRF (MyoclonicEpilepsy withRagged RedFibres).34

Twelve lead electrocardiogram from a patient with Kearns-Sayre syndrome showing the broad QRS complexes of bifasicular block (right bundle branch block with left anterior fasicular block).

Kearns-Sayre syndrome. Note the bilateral ptosis, myopathic facies, and the hearing aid. (Photograph reproduced with permission.)
The second group of patients includes those who have a constellation of clinical features which are highly suggestive of mitochondrial disease, but who do not neatly fall into a specific syndrome category (table 2). Thus, short stature in association with bilateral sensorineural deafness, ophthalmoplegia with ptosis, or a history of migraine and diabetes are three clinical presentations that suggest a mitochondrial aetiology. A meticulous neurological examination is essential in all patients with suspected mitochondrial disease. The combination of any of the above features with clinical evidence of a myopathy and central neurological signs places a mtDNA defect high on the list of differential diagnoses. However, mtDNA disease may mimic other inherited neurological disorders, and patients with mtDNA defects often undergo numerous autosomal genetic tests before the correct diagnosis is made. For example, mitochondrial disease may present as a spinocerebellar syndrome142 and a mtDNA defect should be considered in patients who test negative for the known spinocerebellar ataxia (SCA) mutations (particularly if they have an ophthalmoplegia and if there are no documented paternal transmissions). Focal143 or generalised144 dystonia with optic atrophy may be confused with the early stages of Huntington’s disease. Usher syndrome (pigmentary retinopathy with deafness) shares clinical features with mtDNA disease145 and patients with mtDNA defects may present with a motor and sensory neuropathy similar to Charcot-Marie-Tooth disease.142 Finally, we have seen a number of patients who were intially diagnosed as “congenital myasthenia”. This list of “missed” diagnoses of mitochondrial diseases is by no means complete, but it serves to illustrate the daunting scope of the situation that confronts the clinician.
Mitochondrial respiratory chain diseases
Clinical features of mitochondrial disease
The last of the three main groups is the most difficult to define. Clinicians are seeing an increasing number of patients who are referred with unexplained symptoms and signs who, surprisingly, turn out to have mtDNA disease (table 2). As one example, mtDNA defects cause a small, but significant proportion of strokes in the very young.146 147 Furthermore, mitochondrial diseases often have a non-neurological presentation, with features such as a hypertrophic cardiomyopathy41 43 44 100 148 and renal tubular disease.130 131 149 150 Endocrine abnormalities are common and hypopara thyroidism,29 121 122 151 adrenal insufficiency,152 153 and diabetes99have also been widely documented.154 In fact, between 0.5 and 1% of diabetics harbour a causative mtDNA mutation154and even this prevalence is probably an underestimate because the total number of mtDNA mutations that can cause diabetes is not known. Gastrointestinal abnormalities are also common in mitochondrial patients, although they rarely volunteer this information spontaneously.96 Recurrent vomiting has been mistaken for anorexia nervosa and a gastrectomy was carried out before the correct mitochondrial aetiology was established.72 126Mitochondrial dysphagia may lead to a percutaneous gastrostomy before the age of 2096 and both constipation and diarrhoea are also common. It is unclear at present whether these features result from a visceral myopathy or a myenteric plexus neuropathy.
Although deafness is a common feature of mtDNA disease, one particular mutation deserves a special mention. In a large study of 70 Spanish families with deafness, 27% were found to harbour a homoplasmic point mutation at mtDNA nucleotide 1555. This mutation was associated with both congenital and late onset deafness, and the penetrance was enhanced by exposure to aminoglycosides.105 The identification of this mutation should make it possible to prevent, or at least to delay, the onset of deafness in susceptible subjects by avoiding exposure to aminoglycosides. This problem is potentially of major importance, particularly in countries where streptomycin is frequently used in the treatment of tuberculosis.155
The list of clinical features associated with mtDNA defects is seemingly endless. How then should we approach the problem in the general genetics clinic? It is sometimes possible to elicit a maternal family history, but unfortunately this is often not the case. A history of recurrent spontaneous abortion and early neonatal death is not uncommon in pedigrees of mtDNA disease, but even with an extended geneaology, mitochondrial inheritance may be confused with dominant, recessive, or sex linked inheritance.145 Taking a family history is made all the more difficult when we consider the relatively non-specific features of some mitochondrial diseases. How many families have relatives with diabetes, strokes, heart disease, and migraine? From a practical point of view, it is sensible to consider a mitochondrial aetiology in any case of multisystem disease, particularly if it involves the central nervous system.
In our experience, the overt presentation of mitochondrial disease is less common in children than in adults and it may be quite different. In the neonatal period, mitochondrial disease can present as a metabolic encephalopathy with hepatic and cardiac failure, as well as an associated lactic acidosis.137 Most of these babies die, but it is essential to make an accurate diagnosis because this presentation may mimic X linked pyruvate dehydrogenase complex (PDC) deficiency, or a potentially treatable metabolic disease such as biotinidase deficiency. More accurate diagnosis of a mitochondrial aetiology should thus enable genetic counselling and prenatal diagnosis.156 Older children may present with an encephalopathy (seizures and coma), prominent brain stem signs (bizarre eye movements and ataxia), and extrapyramidal disease (dystonia and chorea) suggestive of Leigh syndrome (also called subacute necrotising encephalopathy). These children often have characteristic neuroimaging on CT, with hypodensities within the basal ganglia.111Leigh syndrome may result from a nuclear gene defect (usually autosomal recessive81 82 85 86), but occasionally X linked PDC deficiency156 or a mtDNA defect (a point mutation in the mitochondrial ATPase6 gene).157 158 Children with mitochondrial disease may also present with sideroblastic anaemia, pancytopenia, and exocrine pancreatic failure (Pearson syndrome).135 Finally, children with mtDNA defects may present with non-specific features such as failure to thrive and developmental delay. It is often difficult to decide when to investigate these patients, and because the most revealing investigations are invasive, other treatable causes should be ruled out before embarking on a series of investigations for mtDNA disease.
The investigation of suspected mitochondrial DNA disease
Mitochondrial medicine is a “young”, but increasingly important clinical specialty. The biochemical and molecular genetic tests help to confirm the clinical diagnosis. It would be both inappropriate and potentially misleading to send samples for mtDNA analysis as part of a diagnostic “fishing exercise”, because the same phenotype can be caused by many different mutations. As one example, at least 11 different pathogenic mutations have been identified in the mitochondrial tRNAleu(UUR) gene, several of which cause MELAS. It is important, therefore, to follow a structured investigatory pathway that is driven by the family history and the findings from clinical examination (fig 5). The investigations fall into two main groups: general investigations that are used to accumulate evidence of the pattern and nature of the different organs involved in mitochondrial disease, and specific investigations that seek biochemical/molecular genetic evidence of a mitochondrial abnormality.
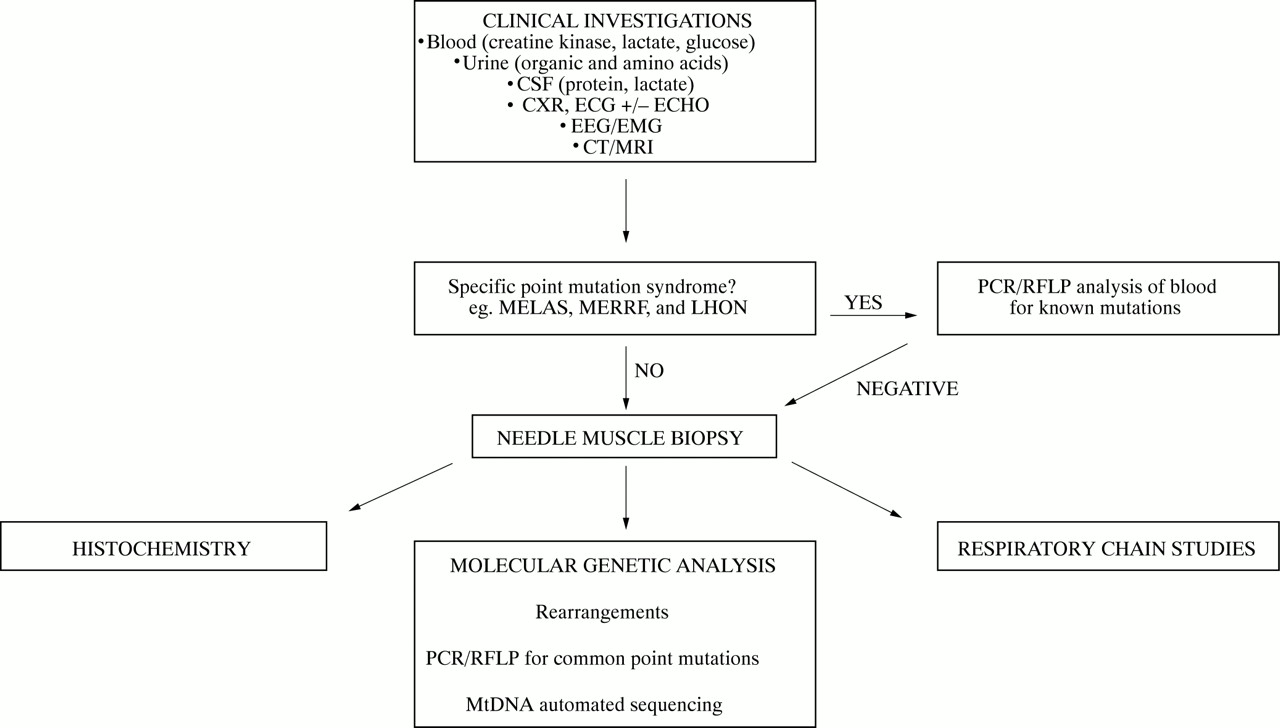
The investigation of mitochondrial DNA disease.
GENERAL CLINICAL INVESTIGATIONS
MtDNA disease is often considered after many other diagnoses have been excluded. For this reason, patients have usually had many general diagnostic tests by the time they are seen by a specialist. It is essential to assess cardiac function (ECG and echocardiography) and to carry out an oral glucose tolerance test because of the high prevalence of cardiac complications and diabetes in patients with mtDNA disease. Patients with a mitochondrial respiratory chain defect sometimes, but not exclusively, have a raised blood lactate level.111 As far as mitochondrial disease goes, therefore, this test is neither specific nor sensitive. There are many causes of a blood lactic acidosis, and many patients with mtDNA defects have a normal blood lactate (particularly when presenting as adults). A raised CSF lactate is a more specific test for mitochondrial disease, if there is central neurological involvement, although many non-mitochondrial neurological conditions result in a CSF lactic acidosis (for example, seizures, migraine, thromboembolic stroke). The serum creatine kinase may be raised, but it is often normal,111 even when the patient has a proximal myopathy. Urine organic and amino acids may also be abnormal in patients with mitochondrial dysfunction.129Every patient with seizures or cognitive decline should have an electroencephalogram (EEG) which may show evidence of seizure activity or diffuse slow waves suggestive of a subacute encephalopathy. They should also have a brain CT scan, which may show calcified basal ganglia (fig 4) or bilateral hypodensities and atrophy, which are valuable diagnostic indicators of mitochondrial disease.159 MRI may also be indicated, particularly in a patient with brain stem signs or stroke-like episodes. The abnormalities are often non-specific (cerebral and cerebellar atrophy),160 161 but they can be indicative of a mtDNA defect.162

CT scan of the brain showing bilateral basal ganglia calcification in a patient harbouring the A3243G (MELAS) mutation.
SPECIFIC INVESTIGATIONS: HISTOCHEMISTRY, BIOCHEMISTRY, AND MOLECULAR GENETICS
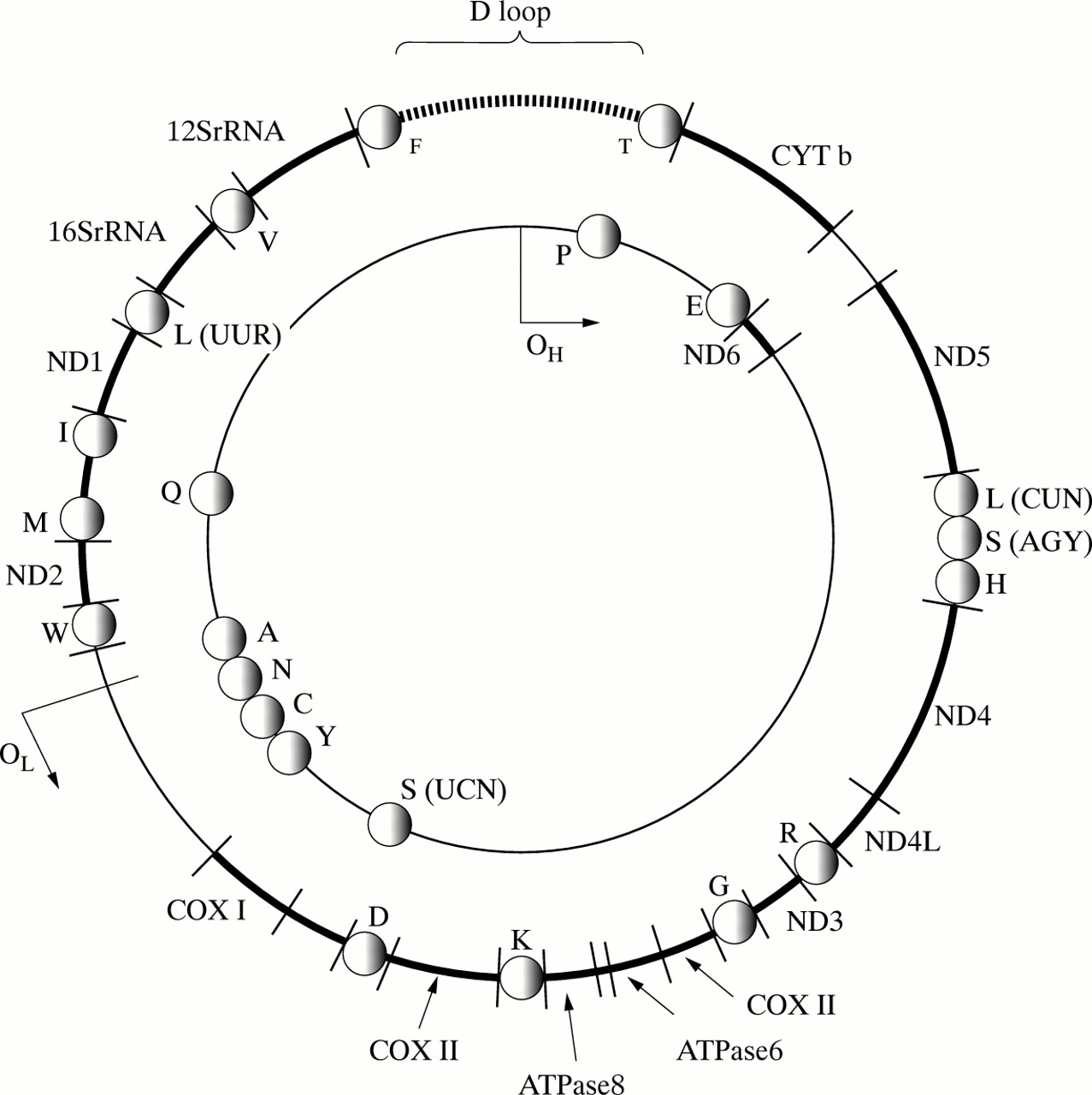
A basic understanding of mitochondrial genetics is essential for the physician who is investigating a patient with suspected mtDNA disease (fig 1). Each human cell contains between 1000 and 100 000 copies of mtDNA. The vast majority of these molecules are identical (homoplasmy) in healthy subjects. Pathogenic mtDNA mutations fall into two groups: rearrangements (deletions and duplications) and point mutations. Patients with pathogenic mtDNA defects usually harbour a mixture of mutated and wild type molecules within each cell (heteroplasmy).93 At the cellular level, the proportion of mutated mtDNA (mutation load) determines phenotypic expression, because the load must exceed a critical threshold level before the cell develops a biochemical defect of the respiratory chain (typically >85% mutated mtDNA).163 Part of the clinical variability associated with a particular mtDNA mutation can be attributed to differences in the overall mutation load among people.117 164 However, the level of heteroplasmy not only varies between people, but it also varies within them. Therefore, unlike nuclear genetic defects which are present within every cell (with the notable, but rare exception of somatic mosaicism), the amount of mutant mtDNA varies from cell to cell and from organ to organ. This complexity provides a further explanation for the diversity of mtDNA diseases, but it can also make the investigation of these patients particularly difficult.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The mitochondrial genome and basic mitochondrial genetics. Human mitochondrial DNA (mtDNA) is a 16.569 bp circle of double stranded DNA that encodes 13 essential respiratory chain protein subunits. The ND1-ND6 and the ND4L genes encode seven complex I subunits (NADH-ubiquinone oxidoreductase), the CYTb gene encodes one complex III subunit (ubiquinol-cytochrome c oxidase oxidoreductase), the COX I to COX III genes encode three complex IV subunits (cytochrome c oxidase), and the ATPase6 and ATPase8 genes encode for complex V subunits (ATP synthase). Interspaced between the protein encoding genes are two ribosomal RNA genes (12S and 16S rRNA), and 22 transfer RNA genes that provide the necessary RNA components for the mitochondrial translation machinery. The protein subunits of the mitochondrial ribosomes are encoded by nuclear genes. The 1.1 kb non-coding region (called the displacement loop or D loop) is important for the generation of mtDNA transcripts and the replication of mtDNA. OH is the origin of heavy (outer) strand replication, OL is the origin of light (inner) strand replication. MtDNA differs from nuclear DNA in a number of ways. (1) Each mononuclear human cell contains between 1000 and 100 000 copies of mtDNA. (2) MtDNA molecules very rarely, if ever, undergo intramolecular recombination. (3) The mitochondrial matrix is rich in mutagens that are generated during electron transfer and oxidative phosphorylation, but mtDNA is not protected by histones, and mitochondria are relatively deficient in DNA repair mechanisms, As a result, mtDNA accumulates mutations at a much faster rate than nuclear DNA. Mitochondrial diseases may result from either mutations of mtDNA or nuclear DNA. Despite the complexities of the mitochondrial genome, its small size makes it amenable to molecular genetic analysis. As a result, over 100 different mtDNA rearrangements and over 50 mtDNA point mutations have been associated with disease. The map position of mtDNA mutations is designated with reference to the L strand of the Cambridge reference sequence.89 Because the tRNA genes are interspersed throughout the genome, mtDNA deletions usually involve a tRNA gene, resulting in a protein translation defect, and causing Kearns-Sayre syndrome, chronic progressive external ophthalmoplegia, or diabetes and deafness (table 1). Point mutations may affect protein coding genes (missense), ribosomal genes, or tRNA genes. Missense mutations may be homoplasmic (all of the mtDNA molecules within the cell are mutated and identical), and cause disorders such as Leber hereditary optic neuropathy (LHON), or neurogenic weakness with ataxia and pigmentary retinopathy through a direct effect on the protein encoding gene. Mutations which involve tRNA genes alter mitochondrial function through a number of different mechanisms that include abnormal transcription90 and abnormal translation.91These mutations result in a wide variety of disorders such as mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), or myoclonic epilepsy with ragged red fibres (MERRF). Patients who harbour these mutations usually have a mixture of mutated and normal mtDNA (heteroplasmy). In vitro studies have shown that a biochemical defect of mitochondrial function is only expressed when a critical percentage level of mutated mtDNA is exceeded. This threshold varies from tissue to tissue and is, in part, related to the dependence of a particular organ upon oxidative metabolism. This variability, coupled with differences in the percentage level of mutated mtDNA within subjects, contributes to the phenotypic variability which is characteristic of mitochondrial tRNA gene disorders. Nuclear gene defects are a major cause of mitochondrial disease, accounting for up to 30% of adult cases and an even higher proportion of paediatric cases. In the majority of subjects, a nuclear gene defect is inferred, whether because of the pattern of respiratory chain involvement, or the distribution of the mitochondrial defect in single muscle fibres. However, nuclear gene mutations have recently been identified in patients with Leigh syndrome resulting from complex I and complex IV deficiency, and point mutations in the succinate dehydrogenase gene cause autosomal dominant optic atrophy and ataxia.
For some mitochondrial diseases, it is possible to obtain an accurate diagnosis with a simple molecular genetic screen (fig 5). For example, 95% of patients with LHON harbour one of three mtDNA point mutations (A11778G,13 A3460G,165 and T14484C of the mtDNA L strand15).166 These patients have very high levels of mutated mtDNA in blood, and it is therefore appropriate to send blood for molecular genetic analysis by PCR/RFLP analysis. Similarly, the level of mutated mtDNA in blood is very high for most MERRF patients who harbour a point mutation in the lysine tRNA gene at position 8344 and, again, the diagnosis can be made from a blood sample.164 In contrast, patients with the A3243G MELAS mutation often have low levels of mutated mtDNA in blood. As a result, it can be estimated that at least 1 in 20 MELAS patients will be missed by the conventional PCR/RFLP approach.167 The mtDNA deletions that cause CPEO and the Kearns-Sayre syndrome are also usually not detectable in blood. The results of such simple mtDNA genetic screens, therefore, must be interpreted with extreme caution. If clinical suspicion was strong enough to warrant the blood test, then patients with a negative result should be investigated further. This usually involves a skeletal muscle biopsy (fig 5). To the unfamiliar, this procedure may seem excessively invasive, particularly if the symptoms and signs are minor. However, needle biopsy is relatively benign and we routinely perform this procedure under local anaesthetic on the neurology ward. The procedure takes approximately 45 minutes from start to finish, is extremely well tolerated, and without major complication (in over 400 biopsies over the last two years).
The analysis of muscle forms the cornerstone of the investigation of patients with suspected mitochondrial disease. Histochemical analysis may show subsarcolemmal accumulations of mitochondria (so called ragged red fibres or RRFs). However, these RRFs are a relatively non-specific finding, because they are seen in a variety of muscle diseases115 and in muscle taken from healthy athletes. Similarly, electron microscopy may show abnormal mitochondria with paracrystalline inclusions but these are also relatively non-specific.168 In contrast, muscle histochemistry may also show cytochrome c oxidase (COX, complex IV) deficient fibres which indicate mitochondrial dysfunction. Respiratory chain complex assays may also show a deficiency.169 Either of these two abnormalities confirm that a patient has mitochondrial disease, and the pattern of the abnormality provides an invaluable guide to the molecular geneticist for additional analyses.
The mitochondrial respiratory chain has an unusual genetic composition (fig 1). Thirteen essential polypeptides are synthesised from genes within the mitochondrion itself, but the majority of polypeptides are synthesised within the cytosol from nuclear gene transcripts. Respiratory chain complexes I, III, IV, and the ATP synthase (complex V) contain both nuclear and mitochondrial subunits, but complex II has a purely nuclear origin. This means that patients with a pure complex II defect must have a nuclear gene defect,83 but patients who have single complex defects that affect I, III, or IV may have a gene defect in either the mitochondrial or the nuclear genome. Adults with mitochondrial disease often have multiple deficiencies in respiratory chain complexes. This finding suggests a more global defect in mitochondrial biogenesis, which is often the result of a mutation involving a mitochondrial transfer RNA gene (fig 1). Further evidence of mtDNA tRNA defect can be gleaned from the histochemistry. Because the level of mutant mtDNA varies from cell to cell (see above), it is often possible to see a mosaic pattern of activity in skeletal muscle, with some fibres positive and some fibres negative for cytochrome c oxidase (COX, complex IV) in these patients.
Southern blotting of genomic DNA, extracted from skeletal muscle, with a range of restriction enzymes will identify duplications and deletions of mtDNA. Common point mutations should be screened by PCR/RFLP followed, as indicated, by direct sequencing of the entire mitochondrial genome. If a heteroplasmic rearrangement, or a recognised pathogenic point mutation is identified, molecular genetic diagnosis is relatively straightforward. Of the 150 patients who we have investigated for possible mitochondrial disease over the last 10 years, a positive finding has been possible in 46% of these cases. However, investigation of the large remainder remains a challenge, and how one should investigate these cases will be the focus of a second article.
Management of patients with mtDNA disease
Although we cannot cure patients with mtDNA disease, optimal management can have a major impact on the level of morbidity and premature death. In broad terms, current management can be divided into three groups: (1) counselling, (2) supportive therapy, and (3) pharmacological therapy. We will then discuss some of the novel treatment strategies which are currently under development and provide some hope for the future.
What does a mtDNA mutation mean for a person?
PROGNOSIS
Clinical and genetic variability are the hallmarks of mtDNA disease. For example, although the A3243G MELAS mutation is found in asymptomatic relatives, it can cause a mild phenotype, such as sensorineural deafness with diabetes, or alternatively it can cause a severe encephalopathy with death at a young age. Because the level of mutant mtDNA determines whether the genetic defect is expressed at the level of the cell, it seems axiomatic that there should be a simple relationship between mutation load and clinical disease. It is only recently, however, that there have been sufficient cumulative data to permit analysis. In a recent study of over 150 patients with the A3243G (MELAS) and A8344G (MERRF) mutations, the level of mutant mtDNA in skeletal muscle was highly correlated with the frequency of the major neurological features associated with each mutation.109 In contrast, however, the clinical features were not related to the level of mutant mtDNA in blood. It appears that, in these patients, high levels of mutant mtDNA are found in postmitotic (non-dividing) tissues such as muscle and neurones, whereas lower levels are found in rapidly dividing tissues such as blood (which remains unaffected clinically). Thus, at least for MELAS and MERRF, the level of mutant mtDNA in a patient’s muscle may give a reasonable guide to prognosis. However, any prognostic advice must be given with caution because the level of mutant mtDNA in muscle may increase65 or decrease170 with time.
PRESYMPTOMATIC TESTING
Over the last few years, we have analysed a number of extended pedigrees that transmit pathogenic mtDNA mutations. As one consequence of these studies, a number of unaffected adult relatives have sought advice about their own prognosis. Because of potential longitudinal changes of mutation load over time, this is a difficult issue. Nonetheless, the available data suggest that, at least for the A3243G and A8344G mutations, the level of mutant mtDNA in muscle may provide a guide to the future. This information must be given with extreme caution, because nuclear genetic and environmental factors may influence the expression of a mtDNA defect.171 172 For these reasons, it is sensible to interpret the molecular genetic data within the context of the family being studied, modifying the estimated risks on the basis of the clinical outcome in other family members and the muscle mutation load of other family members.
INHERITANCE
Although mtDNA deletions themselves are not transmitted,173 174 mtDNA duplications may be transmitted from mothers to offspring.175-178 The duplications themselves are not pathogenic,179 but they predispose to deletion formation.180 It should be stressed that mtDNA duplications are rare, but a female harbouring a mtDNA duplication may thus have an affected child who also harbours a pathogenic mtDNA deletion.
MtDNA point mutations are also inherited down the maternal line.93 For the homoplasmic mutations associated with LHON, extensive pedigree analyses have characterised the risks of blindness in maternal relatives.181 182 However, for heteroplasmic mtDNA point mutations such as those that cause MELAS and MERRF, the situation is more complex. For these diseases, the outcome of pregnancy is dependent both upon the amount of mutant mtDNA transmitted to the oocyte and the segregation of mutant genomes during development. On average, one would expect mothers with high levels of mutant mtDNA to transmit high levels of mutant mtDNA to their offspring. However, analysis of the transmission of the heteroplasmic mtDNA mutations has shown that a simple correlation is not obtained, and that there is a high variability in the mutation load among offspring. A major contribution to this high variability in mtDNA transmission is a restriction/amplification event (called a “bottleneck” by some authors) that occurs during oogenesis.93 Although there have been recent advances in our understanding of the “bottleneck”,183 it is unlikely that the resulting simple mathematical models will be of any practical use in the clinic.184 In the first place, the theoretical “bottleneck” size is calculated from the level of mutant mtDNA in the blood of a mother and her offspring. For the reasons outlined earlier, the level of mutant mtDNA in blood may change with time, and therefore it may not directly reflect the level of mutant mtDNA in the oocyte. Variation in the level of mutant mtDNA in blood may give the impression of an excessively tight “bottleneck”. Secondly, bottleneck models predict that the range of possible outcomes of a pregnancy is huge. For any female, she would be predicted to have offspring harbouring either very low or very high levels of mutant mtDNA.
It is clear that we have a great deal to learn about the mechanism of inheritance of mtDNA mutations and their segregation. Although the “bottleneck” hypothesis may describe the events occurring in vivo, the fine details need to be determined and reliable genetic counselling will only be possible when we have a much more complete understanding of the processes involved in the inheritance, segregation, and expression of mtDNA defects. It was precisely because of these uncertainties that we took a different approach to the problem. Without making any assumptions about mechanism, we studied a large group of mothers who harbour the A3243G MELAS and A8344G MERRF mutations.185 It was found that higher levels of mutant mtDNA in the mother’s blood were associated with an increased frequency of affected offspring, but that the inheritance pattern of the two mutations was different. Although it would currently be unwise to use these data for genetic counselling, the results clearly indicate that the level of maternal mutant mtDNA is important in determining the frequency of clinically affected offspring. With the acquisition of prospective data, it should be possible to improve the genetic advice we give to females in the future.
PRENATAL DIAGNOSIS
The complexities of mtDNA segregation cause difficulties with prenatal diagnosis. For example, the level of mutant mtDNA in a chorionic villus sample (CVS) may be different from the level in the fetus. In isolated cases, the level of mutant mtDNA has been uniformly distributed throughout the fetus, which suggests that a chorionic villus sample might give useful information.186 187However, at present we know little about how the level of mutant mtDNA might change during development, and it will be some time before we know whether the mutation load in the CVS provides clinically useful information.
As far as mtDNA point mutations are concerned, in our experience, mutant mtDNA can be detected in at least some tissues of almost every offspring of a female who harbours a pathogenic mutation, and CVS is thus likely to be positive for all pregnancies of an affected female. At present, we do not know the long term implications of a CVS heteroplasmy measurement, and it would be unwise to counsel purely on this basis. Further studies must be done in this area, but there are considerable ethical difficulties with a procedure that is not without risk.
PHARMACOLOGICAL THERAPY
At present, we have no effective disease modifying treatment for mtDNA disease.188 Various vitamins and cofactors have been used, and varying degrees of success have been reported in individual cases.189-191 The small clinical trials that have been carried out have not shown a consistent clinical effect,192 but, because drugs such as ubiquinone (CoQ10) are relatively free of side effects, there is a rationale for their use in individual cases. In our experience, many patients report a modest improvement, but this is not sustained, suggesting a substantial placebo component to their response.
NOVEL TREATMENT STRATEGIES
Previous attempts at drug or metabolic therapy for mitochondrial diseases have not been encouraging. A moderate degree of exercise has been shown to improve exercise tolerance and muscle metabolism, and this should be recommended to patients with mtDNA disease,193 and some recent laboratory research suggests that real progress may be possible in the near future. Recent in vitro work has shown that sequence specific peptide nucleic acids can inhibit the replication of mutant mtDNA.194 These novel antigenomic molecules can be delivered into human mitochondria in culture, and potentially they may correct a mitochondrial genetic defect in vivo. Secondly, muscle satellite cells are the quiescent precursors of mature skeletal muscle and they contain low levels of mutant mtDNA. The induction of focal muscle cell necrosis induces satellite cell proliferation. In two patients with a mitochondrial myopathy, the subsequent proliferation of satellite cells led to the repopulation of a small region of skeletal muscle with mature muscle that harboured low levels of mutant mtDNA and with normal mitochondrial function.195 196
Conclusion
In this review, we have outlined the common clinical presentations of mtDNA disease, highlighting the importance of a structured, integrated approach to the investigation of these patients. We have discussed what the diagnosis means for the person and described some of the difficulties in giving prognostic advice and genetic counselling. Over the last five years, there has been a vast increase in our understanding of mtDNA disease. This progress has been paralleled by some advances in the clinical management of these patients. However, by the time we come to investigate patients with mtDNA disease, neuronal loss is often extensive and it is unlikely that we will be in a position to reverse the damage that has already been done. In addition, because the level of mutant mtDNA may change with time, it is crucial that we come to understand the mechanisms leading up to the clinical presentation. Although a contentious issue, it is therefore essential for us to study these people before they develop symptoms and signs. In this way, we will eventually be able to give more accurate prognostic information to subjects looking for a presymptomatic diagnosis, and it will be particularly important if we are going to develop disease modifying therapies which prevent the onset of neuronal loss.
Acknowledgments
PFC is a Wellcome Trust Research Fellow. NH acknowledges support from the National Eye Institute (RO1 EY10758), the John Sealy Memorial Endowment Fund, and The Wellcome Trust. RA is a MRC Research Training Fellow. DMT is supported by The Wellcome Trust and the Muscular Dystrophy Group of Great Britain.