Article Text

Abstract
Patients with Tietz syndrome have congenital profound deafness and generalised hypopigmentation, inherited in a fully penetrant autosomal dominant fashion. The pigmentary features and complete penetrance make this syndrome distinct among syndromes with pigmentary anomalies and deafness, which characteristically have patchy depigmentation and variable penetrance. Only one family has been reported with the exact features described in the original report of this syndrome. This family was reascertained and a missense mutation was found in the basic region of the MITFgene in family members with Tietz syndrome. Mutations in other regions of this gene have been found to produce Waardenburg syndrome type 2 (WS2), which also includes pigmentary changes and hearing loss, but in contrast to Tietz syndrome, depigmentation is patchy and hearing loss is variable in WS2.
- Tietz syndrome
- Waardenburg syndrome
- deafness
- MITF
Statistics from Altmetric.com
In 1963, Tietz1 described an autosomal dominant syndrome of hypopigmentation and deafness in a large three generation family (MIM 103500). There were no further reports of this syndrome, and Reed et al 2 suggested the cosegregation of the two features had been coincidental in that family.
We were contacted by a family who suspected they had a unique form of deafness, and they wanted to know if there were any other families with similar findings. In particular, they wanted to locate a branch of their family which had become separated in the mid 1800s. This family has traced their ancestry in the USA to North Carolina in the mid 1700s and believe the founder in the USA emigrated from Derry, Ireland. They described themselves as having profound congenital deafness along with blonde hair and white eyelashes and eyebrows, transmitted through at least seven generations. Review of published reports and comparison with the pedigree information provided by the family indicated that this was a separate branch of the same family reported by Tietz1 (fig 1).
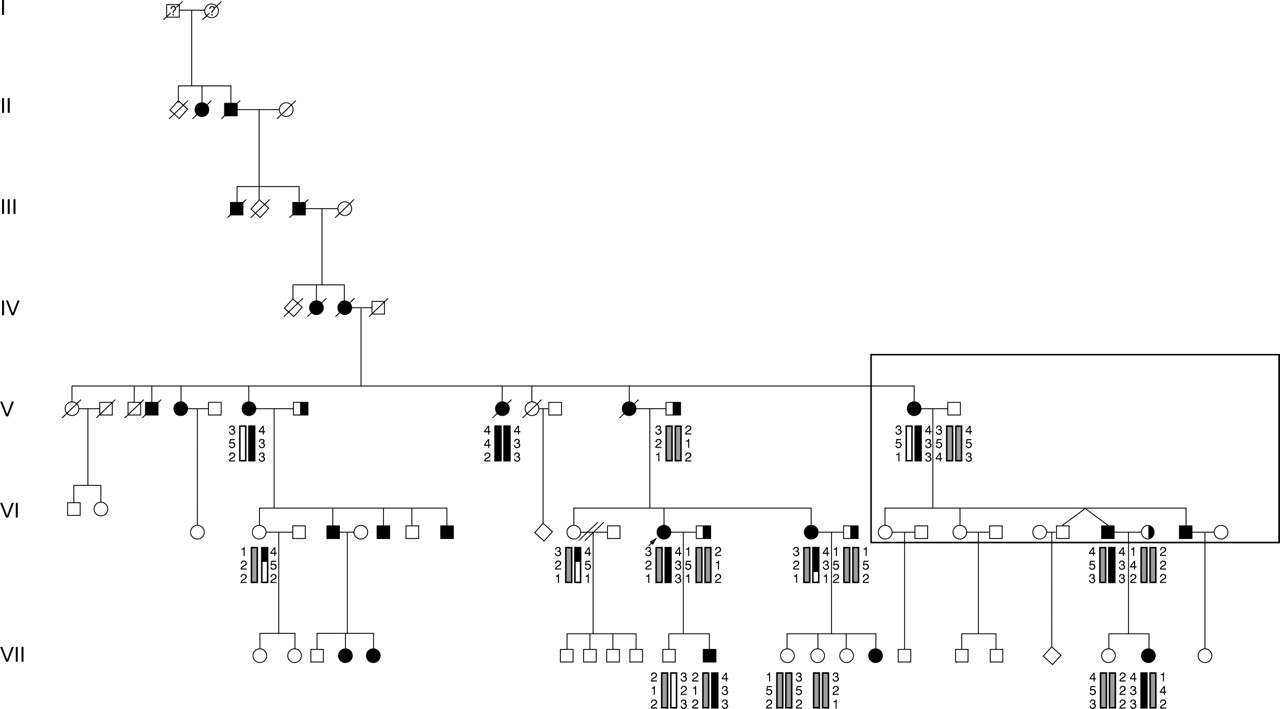
{kind=link}
Pedigree of family 815 showing segregation of the hypopigmentation/deafness through seven generations according to their genealogical records. Filled symbols indicate hypopigmentation and deafness, while half filled symbols indicate isolated deafness. Family members who participated in the 1963 report by Tietz1 are indicated in the box. Haplotypes are shown for markers D3S1285, D3S1566, and D3S1271.
Eleven family members were personally examined and audiograms were obtained on all of them. Six of these 11 family members had more extensive tests at Boys Town National Research Hospital, including vestibular assessment, ophthalmological examination with fundus photography, and physical examination for dysmorphology. Four family members who could not be seen by the authors provided photographs, audiograms, and medical records. Photographs were also available for members of previous generations.
With signed consent, blood was obtained from 19 family members for linkage and mutation analyses. Two point linkage analysis was done using the LINKAGE analysis program with the trait locus treated as a fully penetrant autosomal dominant condition with no sporadic cases.3 This produced a maximum lod score of 3.612 at zero recombination for the marker D3S1566, which is located at 3p12.3. The gene MITF (microphthalmia associated transcription factor) is known to be in this region, and mutations of this gene can produce the Waardenburg syndrome type 2 phenotype.4
Heteroduplex analysis was used for mutation detection inMITF, with direct sequencing to define the mutations. This detected sequence variations in affected subjects in intron 5 and exon 6. The PCR amplimer from exon 5 and adjacent intron sequence included a change of a C to G at nucleotide +9 in the 5′ end of intron 5. This places the mutation out of the 5′ splice consensus region, so that it should not have an effect on the normal maturation of the mRNA. In exon 6, affected subjects were heterozygous for a G to C point mutation at nucleotide 600. This would result in substitution of lysine for asparagine (Asn210Lys) in the mutant protein. These variations were not found in the unaffected subjects, nor in 96 unrelated control samples.
According to family report, affected family members with Tietz syndrome are born “snow white”. They gradually gain some pigmentation and have fair skin and blonde to white hair as adults with white eyebrows and eyelashes. They may have reddish freckles and some subjects reported they can even tan slightly with very careful exposure to sun.
All affected subjects have blue eyes and ophthalmological examination showed hypopigmented fundi, but there is no nystagmus or other visual problems and no heterochromia irides. Hearing loss is always bilateral, congenital, sensorineural, and profound, and affected subjects communicate manually. Vestibular studies showed a slight low frequency phase lead with rotary chair testing on all four tested subjects. This suggests vestibular dysfunction, but caloric testing to delineate the nature of the dysfunction was not performed. Medical records from VI.7 indicated that a CT scan of the temporal bones showed a fistula of the lateral semicircular canal; however, he was a scuba diver, so it is not known if this was related to barotrauma.
Tassabehji et al 5 and Amielet al 6 have described a mother and son who also had a mutation in the basic region ofMITF at the nearby amino acid position 217. This is the only other reported family with a mutation in the basic region, and they also did not have WS2. Both patients were congenitally deaf and the woman had generalised depigmentation with reddish freckles and reddish hair which became grey-blonde at the age of 18. At 18 months of age, her son had auburn hair. Both had lack of retinal pigmentation. Although the degree of hypopigmentation is not identical to the phenotype in the present family, this phenotype was felt by the authors to be closest to Tietz syndrome. The finding of anMITF mutation in our family would confirm that diagnosis.
Waardenburg syndrome type 2 (WS2) is characterised by sensorineural hearing loss of varying degrees in about 70% of gene carriers.7 8 Vestibular dysfunction has been reported in WS2, along with inner ear malformations particularly involving the semicircular canals.9 Depigmentation is patchy, but most typically includes a white forelock, early greying, and heterochromia irides. There is genetic heterogeneity within WS2, in that approximately 15% of families with WS2 have mutations in theMITF gene.7
Although caused by mutation in the same gene which can cause WS2, the Tietz syndrome phenotype is different in several respects. Deafness is always congenital and profound and is completely penetrant, there is no patchiness to the pigmentation, and no heterochromia. Features similar to WS2 include fundus hypopigmentation, possible vestibular dysfunction, and lack of facial dysmorphology.
MITF is a transcription factor of the basic helix-loop-helix zipper family of proteins,10 and appears to be necessary for the development and survival of melanocytes, osteoclasts, and mast cells.10-12 In these transcription factors, the basic portion of the molecule is involved in binding specific DNA promoter sequences, and the helix-loop-helix and zipper motifs stabilise dimerisation with itself or other proteins in the same subfamily. The change in exon 6 of MITF in this family would produce a substitution of lysine for asparagine in the basic region of the transcribed protein. Asparagine is a neutral, polar amino acid, while lysine is basic. Since the basic region of theMITF protein is important for binding to the major groove of DNA, these changes in size and charge would be predicted to interfere with that function. In the heterozygote, the mutant MITF protein would be expected to dimerise effectively with itself or with a normal allele product; however, the resulting dimer would bind to DNA poorly, so the overall effect of the mutation should be dominant negative. In contrast, mutations of MITF that produce WS2 either truncate the product or affect its dimerisation by disrupting the helix-loop-helix or zipper motifs. As shown by in vitro studies of WS2 mutations in the heterozygous state, these mutations do not interfere with the product of the normal allele, which is still free to form a normally functioning protein.12 Thus, the mechanism for WS2 appears to be through haploinsufficiency.6 12 13
MITF is homologous to theMitf (mi) gene in the mouse. Mutations of this gene produce a range of phenotypes including white spotting or pigmentary dilution, inner ear abnormalities, bone resorption problems, and small eyes.14 In mice, there are threemi mutations in the basic region which are inherited in a semidominant fashion, mi,Mior , andMiwh . The mimutation is analogous to the arginine deletion seen in the family with Tietz syndrome reported by Tassabehjiet al 5 and Amielet al 6 and the other two mutations each involve either an asparagine (Miwh, Ile212Asn) or a lysine (Mior , Arg216Lys), indicating that changes involving these amino acids in this critical region have a functional effect. These mutations are felt to act in a dominant negative fashion and produce both dilution of pigmentation and spotting in the heterozygote. A fourth mutation,miew , deletes much of the basic region and also acts as a dominant negative in vitro; however, the phenotype is recessively inherited. It is possible that the defective protein is unable to enter the nucleus and does not dimerise, in effect eliminating the dominant negative action.14 Mutations in other regions in the mouse gene are recessive, with normal phenotypes in the heterozygote despite the reduced levels of functional protein. This is an interesting contrast to the dominant inheritance of WS2, indicating that at least certain tissues in humans (the stria vascularis, irides, and forelock region, for example) are more sensitive to reduced levels of the MITFprotein.4 12 15 16 This sensitivity to amount of protein has also been seen in the hamster. The Wh (anophthalmic white) mutation of the Mitf gene in the Syrian hamster has been reported to produce a white bellied phenotype with overall dilution of pigmentation and variable hearing loss in the heterozygote. This phenotype is the result of a point mutation which truncates the protein in the loop region, interfering with dimerisation. Thus, this mutation appears to act through haploinsufficiency in the heterozygote.17
Yajima et al 18 have called attention to a novel type of mutation in their description of theMitfmi-bw mouse, which is white with black eyes and has severe hearing loss. This phenotype was found to be the result of an insertion of an L1 retrotransposable element in intron 3. This resulted in the decrease in production of twoMitf isoforms, Mitf-A and Mitf-H, and absence of a third isoform, Mitf-M. These isoforms differ in the inclusion of amino acids 5′ to exon 2, and have different tissue distributions, with Mitf-A increased in the retinal pigment epithelium (RPE), Mitf-H predominant in the heart, and Mitf-M increased in melanocytes. Since the RPE is normal in theMitfmi-bw mouse, it was hypothesised that the normal Mitf-A isoform was still present in sufficient amount in that tissue, but that the absence of Mitf-M accounted for the albinism of the skin and the hearing loss. The authors cautioned that similar intronic MITFmutations could exist in human WS2, but that these would be missed by the usual mutation screening of coding regions and splice sites. Furthermore, they noted that two WS2 families had been described with splice site mutations at the exon 1 boundary, causing read through and termination within intron 1. This could have differential effects on the isoforms and thus affect the variable phenotype seen in WS2. However, since Tietz syndrome involves generalised effects on RPE, skin, and the auditory system, it is less likely that differential isoform activity is involved. Additionally, the mutations described for Tietz syndrome have been in the basic region of the molecule, which is common to all three isoforms.
In their 1994 review of MITF mutations, Jackson and Raymond16 noted that the WS2 phenotype appeared to be caused by mutations producing haploinsufficiency ofMITF, and they posed the question as to what the phenotype of a carrier of a dominant negative allele would be. Existing evidence now supports the hypothesis that mutations of the basic region of MITF can act through a dominant negative mechanism, and in at least two cases have produced the congenital deafness and generalised depigmentation characteristic of Tietz syndrome rather than a WS2 phenotype.
Acknowledgments
The authors would like to acknowledge the contribution of Dr G Bradley Schaefer for the dysmorphology examination, Dr David Chacko for ophthalmological evaluation, and Ms Mary Ava Gossman for the vestibular evaluation. We would also like to thank Brian Comer, James Askew, Mike Weston, Larry Overbeck, and Skip Kennedy for technical assistance in this study. Finally, we thank the family involved in this study for their interest, support, and cooperation in the research. The study was supported by grants NIH-NIDCD P60 DC00982 and NIH-NIDCD RO1 DC02942-02 to the first author.
References
Footnotes
-
↵* Present address: Center for Human Molecular Genetics, Munroe Meyer Institute, 985455 University of Nebraska Medical Center, Omaha, NE 68198-5455, USA