Article Text

Abstract
Introduction: Primary open-angle glaucoma (POAG) is a leading cause of visual impairment worldwide and a complex genetic disorder that affects mostly adults. Mutations in the MYOCILIN (MYOC) and OPTINEURIN genes account for rare forms with a Mendelian inheritance and for <5% of all POAG cases. The CYP1B1 gene, a member of the cytochrome P450 gene family, is a major cause of primary congenital glaucoma (PCG), a rare and severely blinding disease with recessive inheritance. However, CYP1B1 mutations have also been associated with cases of juvenile-onset glaucoma in some PCG families or shown to modify the age of onset of glaucoma linked to a MYOC mutation in a large family.
Objective: To investigate the role of CYP1B1 mutations in POAG predisposition, irrespective of the presence of a MYOC mutation.
Methods and subjects:CYP1B1 coding region variation was characterised by denaturing high performance liquid chromatography (DHPLC) and sequencing in 236 unrelated French Caucasian POAG patients and 47 population-matched controls.
Results: Eleven (4.6%) patients carried one or two mutated CYP1B1 gene(s) and no MYOC mutation. They showed juvenile or middle-age onset of disease (median age at diagnosis, 40 years, range 13–52), significantly earlier than in non-carrier patients. Apart from one, all mutations detected in POAG patients were previously associated with PCG.
Conclusion:CYP1B1 mutations might pose a significant risk for early-onset POAG and might also modify glaucoma phenotype in patients who do not carry a MYOC mutation.
- DHPLC, denaturing high performance liquid chromatography
- DMSO, dimethylsulfoxide
- IOP, intraocular pressure
- PCG, primary congenital glaucoma
- POAG, primary open-angle glaucoma
- TEAA, triethylammonium acetate
- CYP1B1
- genetics
- glaucoma
- mutation
- screening
Statistics from Altmetric.com
- DHPLC, denaturing high performance liquid chromatography
- DMSO, dimethylsulfoxide
- IOP, intraocular pressure
- PCG, primary congenital glaucoma
- POAG, primary open-angle glaucoma
- TEAA, triethylammonium acetate
Primary open-angle glaucoma (POAG) is an optic neuropathy characterised by an excavation of the optic disk and a progressive alteration of the visual field. It is a leading cause of visual impairment, affecting 66 million persons worldwide.1,2 Genetic factors play a well-established but complex role in POAG predisposition.3,4 Six loci with a Mendelian inheritance have been mapped using large multicase families.5 Two of these, including GLC1A/MYOCILIN (MYOC) and GLC1E/OPTINEURIN (OPTN), have been identified at the molecular level.6,7 In most cases, however, and in spite of clear familial clustering, POAG does not follow a Mendelian pattern of inheritance. Mutations of MYOC have been detected in a minor (2–4%) proportion of the most frequent cases.8 Altogether, the genetic basis of POAG remains unknown in the majority of patients. Identifying genetic factors associated with this prevalent disease is nonetheless of substantial research and clinical importance.
Primary congenital glaucoma (PCG) is a rare ocular disorder characterised by marked elevation of intraocular pressure (IOP) at birth or in early childhood, leading to ocular enlargement (buphthalmos) and corneal oedema.9 If it is not urgently treated, optic nerves are irreversibly damaged and vision may be lost. This severe defect is mostly inherited in an autosomal recessive manner.10 A major gene, CYP1B1, belonging to the superfamily of cytochromes P450 and mapping on chr.2p21, has been identified.11,12 A broad diversity of mutations in this gene has been characterised in different populations.13–19
PCG is currently considered as a consequence of a developmental defect of the iridocorneal angle and a clinical and pathogenetic entity distinct from POAG.9 In certain pedigrees, however, both PCG and POAG segregate, suggesting that shared or overlapping mechanisms might predispose to both forms of glaucoma. The identification of CYP1B1 provides an opportunity to test this hypothesis. Indeed, three studies have reported delayed expression of a CYP1B1 mutation and co-existence of PCG and POAG in the same pedigree.1,20,21 Moreover, in a large family where MYOC-linked POAG segregated, a heterozygous mutation of CYP1B1 was associated with earlier onset of the disease in patients carrying the MYOC mutation, indicating that a CYP1B1 mutation might behave as a modifier of the MYOC gene.21
Considered collectively, these observations suggested that mutations of CYP1B1 might be a cause of POAG or at least a risk factor for this disease. In the present study, we report two additional pedigrees with both POAG and PCG patients carrying CYP1B1 mutations. This led us to investigate CYP1B1 mutations in a group of 236 French patients with POAG.
METHODS
Patients
POAG patients were recruited at the Glaucoma Institute of Saint-Joseph Hospital and at the Quinze-Vingts Hospital in Paris, and at the Lille University Hospital as described.22 Probands of PCG families were reported previously.18 A protocol approved by the Ethics Committee of Necker Medical School and in keeping with European legislation was followed. Written informed consent was obtained from all patients or, for children, from their parents. Ophthalmologic examination included slit-lamp biomicroscopy, optic nerve examination, measurement of IOP by applanation tonometry, and visual field assessment by automated perimetry. POAG was defined by the conjunction of a normally open iridocorneal angle, characteristic cupping of the optic disk, and alteration of the visual field. Patients with cataract or media opacities and those with a cause of secondary glaucoma, notably including exfoliation, pigment dispersion, history of trauma, surgery, and glucocorticoid exposure, were excluded. Elevation of IOP was not an inclusion criterion. Final POAG diagnosis was made at the time of inclusion after review of inclusion and exclusion criteria, and before genotyping was conducted. Genotyping was performed blind to clinical data. All patients had been screened for MYOCILIN mutations by denaturing high performance liquid chromatography (DHPLC). Seventeen of these patients carried a MYOC mutation.22
CYP1B1 mutation search
Two methods were employed when searching for mutations in CYP1B1 (GenBank accession number: U56438). First, most of the coding region, including exon II and the 3′ half of the coding region of exon III, could be screened by denaturing high performance liquid chromatography (DHPLC). Pilot experiments indicated that all the mutations that had been characterised in PCG patients from France,18 were reliably detected by this method. Overlapping fragments ranging from 302 to 456 base pairs were amplified by PCR using the primers and conditions shown in table 1. The PCR was performed in a 25 µl mixture containing 100 ng of genomic DNA, 0.4 µmol/l of forward and reverse primers, 1.5 mmol/l MgCl2, 5% dimethylsulfoxide (DMSO), 200 µmol/l of each dNTP, and 0.5 U of Taq DNA polymerase (Invitrogen, Life Technologies, Carlsbad, CA, USA). Cycling conditions for each cycle were: 1 min at 94°C, 1 min at an annealing temperature specific to each amplicon (shown in table 1), and 1 min at 72°C, for 35 cycles. DHPLC analysis was conducted with the Wave Nucleic Acid Fragment Analysis System (Transgenomic, Omaha, NE). The oven temperature and gradient conditions for heteroduplex detection were predicted with WaveMaker software (version 4.1.40) and specified experimentally as described previously.22 Actual parameters for DHPLC analysis of CYP1B1 amplicons were established in this work and are listed in table 1. PCR products from DNA to be tested were mixed with an equimolar amount of PCR product from a DNA sample known to be unmutated. This DNA carried the common SNPs forming the most frequent haplotype in the French population (C3947G, G4160T, C8131G, C8184T, A8195G). The mixture was heated at 95°C for 5 min and allowed to cool at room temperature for 30 min to form heteroduplexes. A 5 µl sample was loaded on a DNA Sep Column (Model L-7300+, Transgenomic) and was eluted with a linear gradient of 5% triethylammonium acetate (TEAA) (buffer A) and 25% acetonitrile+5% TEAA (buffer B) at a flow rate of 0.9 ml/min. Elution profiles were compared to those of normal DNA. Samples with altered profiles were considered as potentially having a sequence variation and were sequenced bidirectionally as previously.13,14 Representative profiles are shown in the online supplemental fig 1 (available at http://jmg.bmjjournals.com/supplemental/).
Primer sequences of CYP1B1 amplicons, PCR conditions, and DHPLC analysis parameters

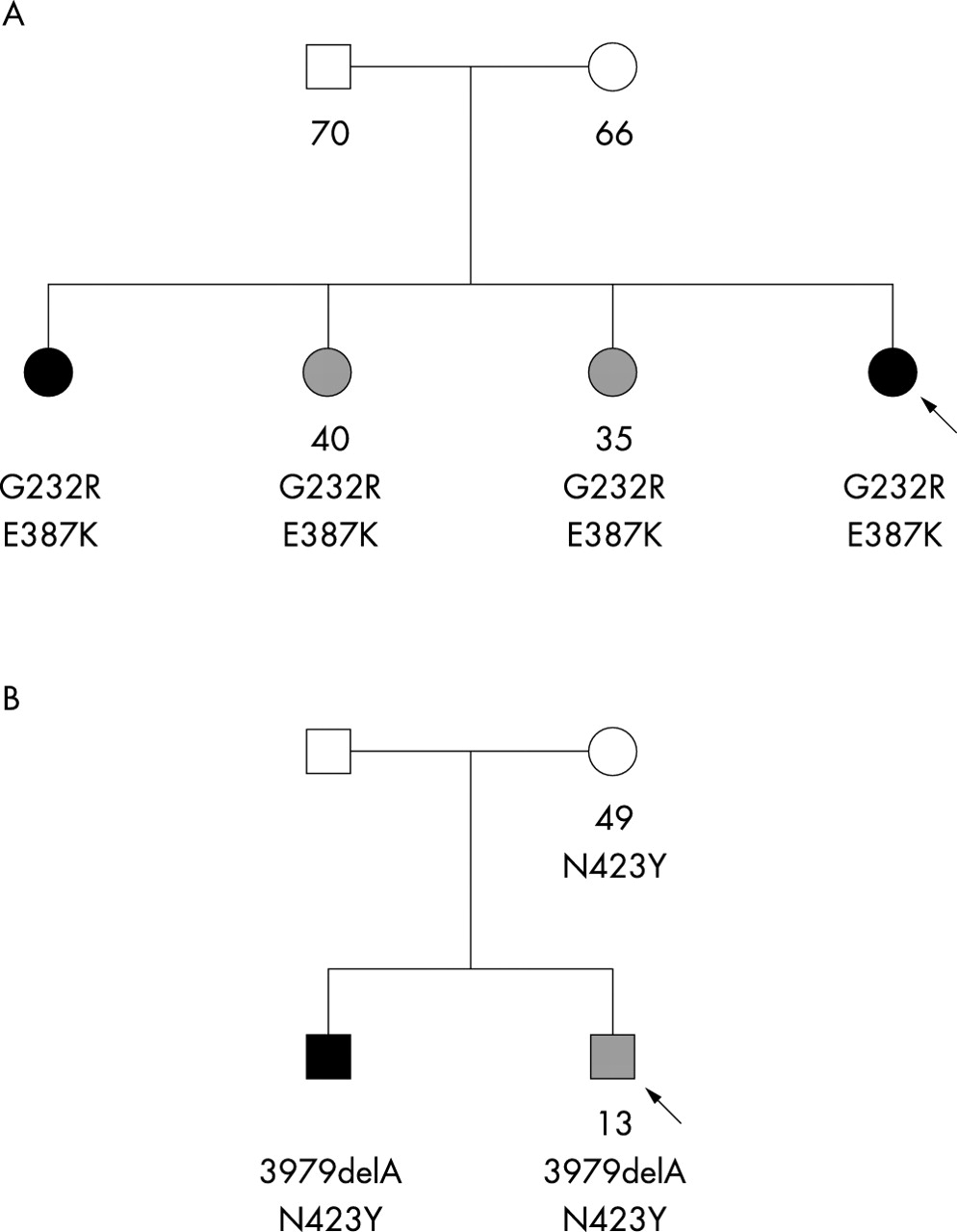
Variable expressivity of CYP1B1 mutations in two sibships. The arrows show the probands. Available genotypes and ages at diagnosis or at examination are indicated below the symbols. (A) Four sisters, all carrying two missense mutations, G232R and E387K. Two were affected with PCG (dark symbol) while the other two developed POAG (hatched symbol). The parents, who are obligate carriers, showed no glaucoma symptom at the indicated ages. Because they could not be genotyped, the phase of both mutations is unknown. (B) Two brothers, both carrying a deletion mutation (3979delA) and a missense mutation (N423Y). The proband developed juvenile glaucoma at the age of 13, while his younger brother showed PCG. Their mother carried the missense mutation and showed no glaucoma symptom at the age of 49. Their father could not be examined.
In contrast, the 5′ end of exon III was not amenable to DHPLC analysis. This DNA segment contains three common SNPs. Their various combinations generate a diversity of elution profiles that complicates the detection of mutations. The corresponding amplicon was therefore sequenced directly.
RESULTS
POAG and PCG in two French families with CYP1B1 mutations
In the pedigree shown in fig 1A, two sisters, including the proband, were affected with PCG whereas the other two sisters developed POAG at ages 35 and 40. The four sisters carried the same two mutations of CYP1B1, G232R and E387K. Similarly, in the pedigree shown in fig 1B, the proband developed juvenile-onset glaucoma at the age of 13. Subsequently, it was found that his younger brother was affected with PCG, which led to investigation of CYP1B1 in this family. Both brothers carried the same two mutations, 3979delA and N423Y.
CYP1B1 mutation screen in 236 unrelated POAG patients
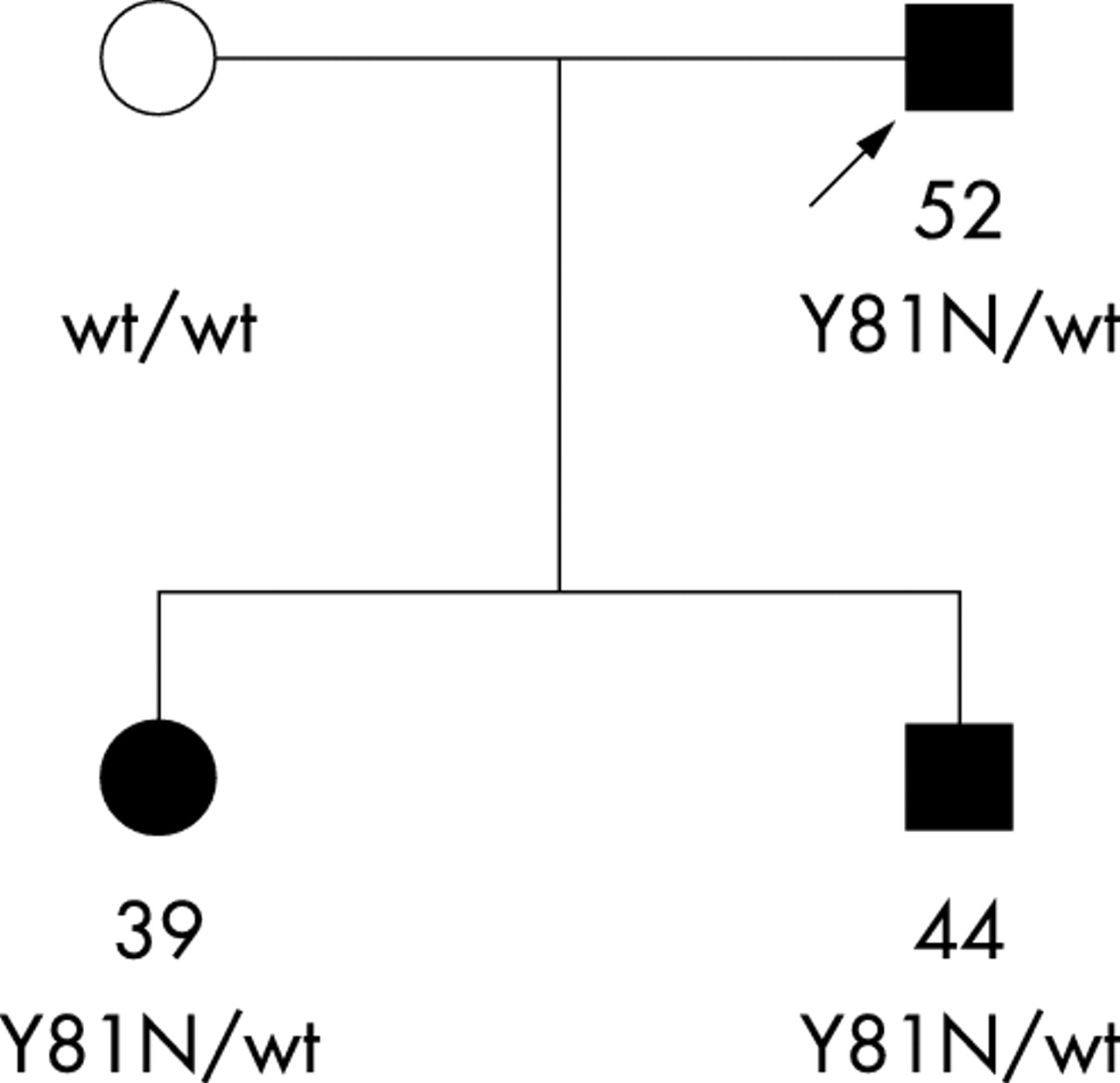
Sequence variation in the CYP1B1 gene coding region was then investigated in a group of 236 unrelated Caucasian patients with POAG from France. Altogether, 11 patients carried a mutation of CYP1B1 (table 2). None of them carried a MYOCILIN mutation. One of the CYP1B1 mutations, Y81N, is new. It was present in the heterozygous state in two unrelated patients. It is likely to be pathogenic because the amino acid change is non-conservative while the tyrosine residue at this position is highly conserved across species in the CYP1 gene family and even among other members of the cytochrome P450 superfamily (see online supplemental fig 2, available at http://jmg.bmjjournals.com/supplemental/). In addition, one of the two patients (patient C) was the father of two children who developed early-onset glaucoma, with ages at onset of 39 and 44 and who both carried the mutation (fig 2). Finally, the mutation was not detected in 394 control chromosomes (see below).
CYP1B1 mutations and rare variants in 12 among 236 unrelated French POAG patients and their clinical initial features at the time of diagnosis
{kind=link}
{kind=link}
Pedigree showing cosegregation of the heterozygous Y81N CYP1B1 mutation with POAG (solid symbols). The arrow shows the proband. Available genotypes and ages at diagnosis are indicated below the symbols.
All other mutations encountered in this group of POAG patients were previously observed in PCG patients, in France or elsewhere. The patient A with juvenile-onset glaucoma and with a compound heterozygous mutation (table 2) was described above (fig 1B). He was included as he was the proband of his family. For all other patients, no family history of PCG was recorded. In particular, they were unrelated to French PCG patients previously studied.18 One mutation, 269delSNF, was homozygous. The carrier was of French descent, whereas the mutation was initially described in PCG patients from Saudi Arabia and, in France, in families originating from North Africa.18,23 All other mutations were heterozygous. The heterozygous E229K mutation was previously associated with PCG in two French cases.18 Two other mutations, including 7901_7913delGAGTGCAGGCAGA (two cases) and R390H (one case), were previously associated with PCG in conjunction with a CYP1B1 mutation on the other chromosome.13,18 The last mutation, A443G, has been observed in Caucasian PCG patients from different countries.15,17 In the three French POAG patients, it was associated with one or two rare synonymous variants, including G4534C (V243V) and T7915C (L360L). The latter variant has not been described before. The A443G mutation has not been observed in Caucasian controls.13,14 Interestingly, a recent report indicated that it could be a common variant in an African population, with a frequency of 7%.24 The study, however, included no assessment of vision while POAG is more frequent (5–10%) and occurs earlier in black than in white subjects.2,3 With this caveat, the pathogenic property of A443G might be population-specific, perhaps because it depends on other factors, themselves population-specific. Alternatively, the mutation might be in linkage disequilibrium with an actually pathogenic variant in Caucasians.
Besides the above described mutations, the six common SNPs previously reported including T3793C, R48G, A119S, V432L, D449S, and N453S were also observed in POAG patients.14,17,18,23 None of these SNPs nor a particular haplotype formed by them was preferentially associated with the mutations (not shown). The rare synonymous variant G4534C (V243V) was detected by itself in one case. No other CYP1B1 variant was detected in the POAG group. Previous reports of Caucasian controls totalling 150 persons from three populations, in the United Kingdom, Turkey, and Saudi Arabia, indicated an absence of CYP1B1 variants apart from the six common SNPs.13,14 We also searched for CYP1B1 variants in 47 French Caucasian controls who were spouses belonging to large families of POAG patients carrying a MYOCILIN mutation. Based on gonioscopy, on assessment of the optic disks, and on an IOP measurement in the normal range, their ophthalmic examination was estimated to be normal. The mutation screening protocol was exactly the same as that used for the group of patients. One mutation, R368H, which was previously associated with PCG,17,21 was detected on one chromosome (table 3). Three synonymous variants, including C3851T (L16L) (one case), C4369A (G188G) (two cases), and V243V (two cases) were also found.
CYP1B1 mutation and variants in 47 French controls
Altogether, the finding of 11 mutation carriers among 236 patients (4.6%) compared to one carrier in 197 controls (47 French+150 previously reported controls) is significant (two-sided exact p = 0.008). Also, if one considers a prevalence of 2.85×10-5 for PCG cases in Western countries,25 the expected frequency for heterozygotes is approximately 1%, assuming a recessive mode of inheritance and under the condition of Hardy-Weinberg equilibrium. The finding of 11 mutation carriers among 236 subjects is significant (two-sided p value using a binomial law = 0.0001, with the null hypothesis of a probability value of 0.01). The calculation, however, is conservative because one of the POAG patients had a homozygous mutation while the heterozygous E229K mutation is associated with PCG in France and also because all PCG cases are assumed to be caused by CYP1B1 mutations.
Clinical features of CYP1B1 carriers with POAG
All patients listed in table 2 had definite bilateral POAG with cupping of their optic discs and significant alteration of their visual field. Evolution of glaucoma was severe enough to require either surgical therapy on both eyes (four patients), argon laser trabeculoplasty on both eyes (one patient), or surgical therapy on one eye and medical bi- or tri-therapy on the other eye (four patients). Of note, their iridocorneal angle was recorded as being normal. The most remarkable feature was their young age at the time of diagnosis, with a median of 40 years (range: 13–52). Epidemiologic studies indicate that the prevalence of POAG is 5–10 times lower before the age of 60 compared to that after this age.3,26 Therefore, CYP1B1 mutations were typically associated with juvenile and middle-age onset POAG. Moreover, although the age at diagnosis in the whole group of patients also was young (median: 48; range: 6–81; interquartile range: 37–58), the age at diagnosis of CYP1B1 carriers was nonetheless younger than that of non-carriers (p = 0.023, Mann-Whitney test).
DISCUSSION
Our data demonstrate an increased prevalence of CYP1B1 mutations among French POAG patients. They extend the previous findings of Vincent et al,21 indicating that mutations in the CYP1B1 gene, nearly all of which were previously associated with PCG, are a significant risk factor for early-onset POAG also in patients who do not carry a MYOC mutation. Because parents of PCG patients, who carry heterozygous mutations of CYP1B1, are apparently not at increased risk of developing glaucoma (see Bejjani et al23 and this work), gene dosage is unlikely to be a general mechanism for delayed expression of heterozygous mutations. Additional factors are probably necessary for development of the glaucomatous process. Apart from a mutation of the MYOC gene, a modifier locus influencing penetrance of CYP1B1 mutations has been described.23 This locus or a similar one could explain the delayed onset of disease in patients with a homozygous mutation or in compound heterozygotes. Moreover, in an experimental mouse model with a knock-out cyp1b1 gene, a recent report indicated that an alteration of the dopamine pathway could markedly exacerbate mild ocular symptoms associated with a defective cyp1b1 gene.27 Alternatively, one of the hallmarks of the CYP1B1 gene is its inducibility by xenobiotics and by mutagenic chemicals.28 Therefore, onset of glaucoma in CYP1B1 mutation carriers might be caused by exposure to an environmental factor of this kind.
Intriguingly, the IOPs of CYP1B1 mutation carriers at the time of diagnosis were variable, ranging from borderline (20 mm Hg) to markedly elevated (40 mm Hg). Given that marked elevation of IOP is a prominent feature of PCG, the observation of normal to moderately elevated IOP measurements was unexpected. However, the recent discovery of expression of CYP1B1 in the posterior segment of the eye, notably in the neuroretina,29 strongly suggests that mechanisms that do not depend directly on IOP elevation may also explain a role for CYP1B1 in glaucoma pathogenesis. For example, accumulation of a toxic substrate as a result of a defective CYP1B1 gene would result in greater sensitivity of neural ganglial cells to other stress factors and notably would lower the threshold necessary for IOP to cause damage to the optic nerve.
Although mutations of the CYP1B1 gene have been identified in PCG patients from different populations, the frequency of the mutations, their nature, and their diversity vary greatly with the population studied. Similarly, the role of MYOC mutations in POAG appears to very much depend on the ethnic origin of the patients.22,30 It will be therefore important to reproduce our findings in different groups of patients, considering both clinical and population factors in their selection, perhaps by means of a multinational collaborative study.
Identifying CYP1B1 mutations in persons at risk of developing glaucoma, particularly among relatives of POAG patients, is of considerable clinical interest. It should help to monitor vision and to detect early symptoms of POAG in these persons. Because simple treatments are already available and operate optimally when they are given at an early stage of the disease, mutation screening for CYP1B1 could contribute to preventing the visual impairment caused by glaucoma. Finally, common polymorphisms of the CYP1B1 gene have been reproducibly associated with varying risks of developing several cancers,31,32 whereas in mice inactivation of cyp1b1 was associated with an increased resistance to chemically-induced lymphomas.33 The observation of subjects with null alleles of CYP1B1 therefore should provide invaluable insight into the role of CYP1B1 in carcinogenesis34 as well as in other physiological processes involving this gene in the human species.
Acknowledgments
We are grateful to Dr Françoise Valtot, Dr Jean Claude Dascotte, and Professor Alain Béchetoille for providing access to the clinical records of their patients.
REFERENCES
Footnotes
-
This work was supported by INSERM (Institut National de la Santé et de la Recherche Médicale), by the Assistance Publique des Hôpitaux de Paris (grant DRRC-AOM96110), by the Fondation pour la Recherche Médicale, and by Insite Vision Inc. (Alameda, CA). RM was awarded a fellowship of the Comite Mixte Inter-Universitaire Franco Marocain (AI 237/SVS/2000).
-
Conflict of interest: none declared.