Article Text

Statistics from Altmetric.com
Recent advances in molecular cell biology have revolutionised our understanding of medical diseases and provided new and alternative strategies for developing treatments. Here we review the spectrum of molecular therapies that are either currently available or have potential application as agents that are able to modulate the wound healing response in the eye. For the purposes of this review, we define molecular therapy as the targeting of specific molecules known to be involved in the processes of wound healing. This may be at the level of either protein or gene expression.
Ocular wound healing
The process of wound healing is involved in either the pathogenesis or failure of treatment of many of the major blinding or visually disabling conditions in the world today. It is implicated in scarring diseases throughout the eye, some examples of which are described below.
The conjunctival wound healing response is important in many ocular conditions such as pemphigoid, trachoma, and chronic cicatrisation, where the development of complications arises from the disruption of the ocular surface.1 It is also a major determinant of outcome following glaucoma filtration and squint surgery.2The severity and extent of clinical disease are closely related to the degree of conjunctival scarring. Another example of scarring is that occurring in the cornea after excimer laser photorefractive keratectomy (PRK), giving rise to symptoms of haze and resulting, in some cases, in a reduction of the best corrected visual acuity.3 4 The scarring condition of proliferative vitreoretinopathy (PVR) accounts for 7–10% of surgical failures in primary retinal detachment repair procedures5 6 and is responsible for producing significant visual morbidity and blindness. It is characterised by the development of fibrosing and proliferating cellular membranes on the vitreous and retinal surfaces that contract and cause irreparable tractional retinal detachments.
Molecular and cellular events in wound healing
The complex process of wound healing may be simplified by dividing the sequence of events into three phases: the inflammatory, proliferative, and the remodelling phases.7 The inflammatory phase is characterised by the influx of neutrophils and monocytes to the wound area followed by lymphocytes and macrophages, and the release of a variety of growth factors and other molecules. Re-epithelialisation, and an increase in fibroblast activity and number occur in the proliferative phase, as also does angiogenesis and the laying down of granulation tissue. The remodelling phase consists of fibroblasts mediating the processes of wound contraction, extracellular matrix deposition, degradation and modification (that can last several months), and producing regulating growth factors.
The processes involved in the scarring response are under the control of a number of molecules, especially growth factors. Growth factors, as well as the growth factor receptors through which their effects are mediated, present themselves as potential targets for therapeutic intervention. The transforming growth factor beta (TGF-β) superfamily, in particular, has been shown to play a pivotal role in scarring throughout the body, where it is believed to be a potent stimulator of scarring.8-10 TGF-β has also been implicated as a potent stimulant of the scarring process in the eye.11-14 Its actions on stimulating fibroblast functions during wound healing occur via its binding to specific cell surface protein receptors14—namely, TGF-β receptor types I, II, and III.
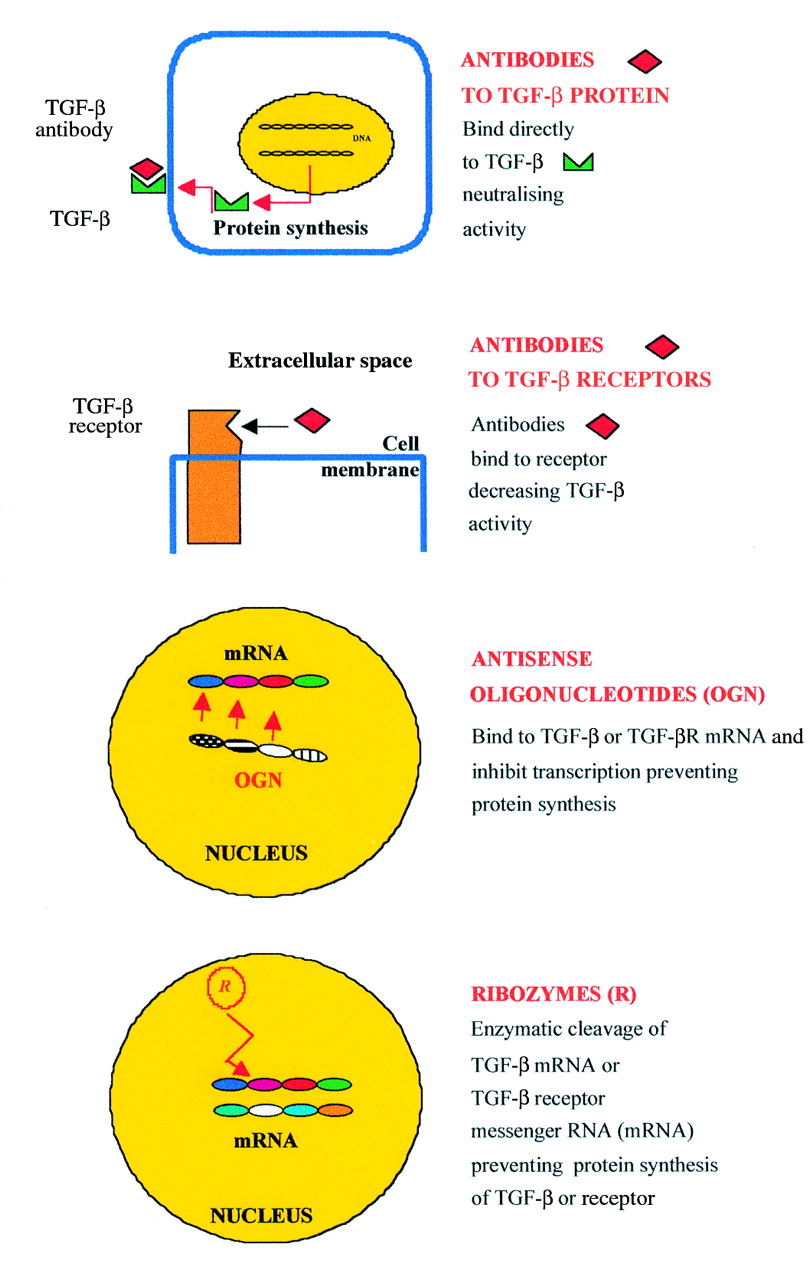
In this review, we use TGF-β as an example of a target for molecular therapy and the modulation of wound healing in the eye. Figure 1illustrates various mechanisms in which the actions of TGF-β may be antagonised. Therapy may be directed against either the TGF-β receptors (TGF-βR type I or II) or their ligands (TGF-β1, β2, or β3), at protein or mRNA levels. The role of fully human neutralising monoclonal antibodies, gene therapy, antisense oligonucleotides, and ribozymes is discussed below.

Targets for molecular therapy. TGF-β activity can be inhibited in several ways using different molecular approaches. Therapy may be directed against TGF-β and receptor proteins using fully human neutralising monoclonal antibodies. Alternatively, inhibition of TGF-β and receptors at the level of gene expression is possible using antisense oligonucleotides or ribozymes, which act at the level of messenger RNA (mRNA) and prevent protein synthesis.
Fully human neutralising monoclonal antibodies
Fully human neutralising antibodies represent a major advance in the field of biotechnology. The term “neutralising” is used to emphasise that on binding of the antibody, the activity of the target antigen is effectively and significantly inhibited. Monoclonal as opposed to polyclonal antibodies are the product of a single clone of B lymphocytes.
Historically, monoclonal antibodies (mAb) were obtained from mice, where the splenic cells from an immunised animal were fused with cells of a cultured myeloma cell line to form a hybridoma that was capable of synthesising one single antibody.15 Unfortunately, murine monoclonal antibodies invariably induced the so called human anti-mouse antibody (HAMA) immune response, a limiting factor in developing therapeutic applications. Attempts at reducing the HAMA immunogenic response led to the development of “chimaeric” antibodies (approximately 70% human protein sequences) and, more recently, the construction of “humanised” engineered antibodies, achieved by grafting the murine antigen binding or hypervariable region on to a human antibody (95% human protein sequence).
However, the production of fully human monoclonal antibodies (100% human sequence) is the most promising advance in this area. Such antibodies can now be produced by two different techniques using transgenic mice and phage display technology. Transgenic mice are genetically modified so that they express human antibody genes. If the antigen is immunogenic, following immunisation these animals produce fully human mAb.16 The technique of phage display consists of three steps: firstly, human antibody light and heavy chains are identified from a large human antibody library; secondly, the human genes coding for these chains are incorporated into a bacterial virus (phage) (Fig 2A); finally, when the phage infects a bacterium (usually,Escherichia coli), the bacterium makes the antibody protein which it displays on its surface (Fig2B).17 18 A phage antibody is a bacterial virus that is engineered to display antibody proteins which will bind specifically to a target molecule. Specific clones binding to an antigen can then be amplified and used to produce the antibody fragment inE coli or other suitable organisms so that a fully human recombinant mAb is produced (Fig 2C).
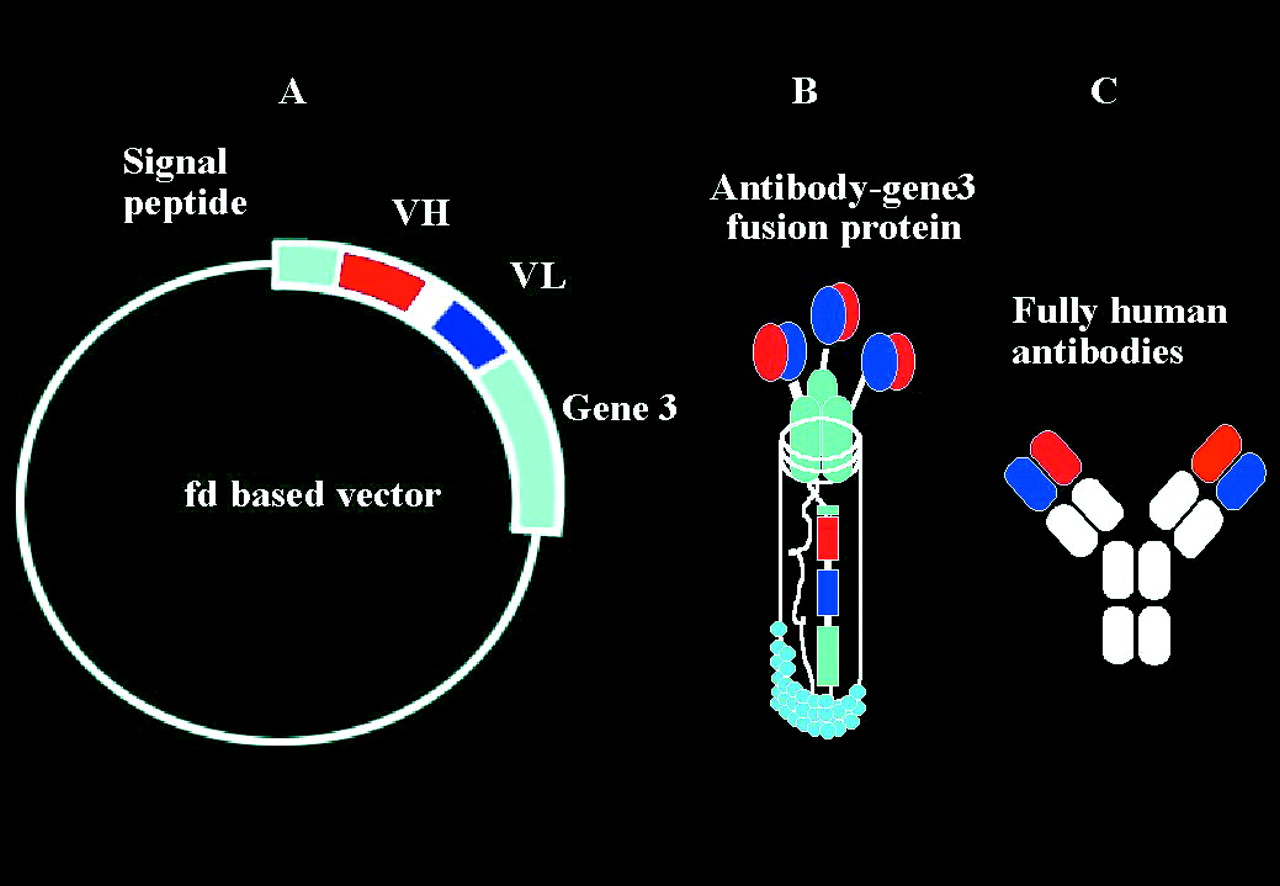
Phage antibodies. Fully human neutralising monoclonal antibodies can be made using the technique of phage display (Cambridge Antibody Technology, Melbourne UK). Human antibody light and heavy chains are identified from a large human antibody library. The human genes coding for these chains are then incorporated into a bacterial virus (phage) (A). When the phage infects a bacterium (usually, E coli), the bacterium makes the antibody protein which it displays on its surface (B). Specific clones binding to an antigen can then be amplified, and the whole antibody produced using recombinant methods (C).
Using the technique of phage display, CAT (Cambridge Antibody Technology, Melbourne UK) has produced a mAb that is specific to the active form of human TGF-β2 - rhAnti-TGF-β2 mAb. Preclinical studies in our laboratory showed efficacy of the antibody both in vitro and in vivo in reducing conjunctival scarring.19 Not only was rhAnti-TGF-β2 mAb found to effectively inhibit human Tenon fibroblasts in several functional assays at IC50concentrations of less than 1.0 nM, but in an animal model of aggressive conjunctival scarring,20 it significantly improved glaucoma filtration surgery outcome compared with control (p<0.03), and was clinically safe, non-toxic, and well tolerated on subconjunctival administration. Interestingly, when compared with the effects of mitomycin C treatment histologically, rhAnti-TGF-β2 mAb appeared much less destructive to local tissue.19
A phase I clinical trial looking at rhAnti-TGF-β2 mAb in primary glaucoma filtration surgery is now completed. At the time of writing, the treatment blind is unbroken, but no serious adverse effects have been noted, and there is evidence of good tolerance and safety. A phase II trial is due to start in autumn 1999. We are also involved with in vitro studies of another antibody—anti-TGF-β1 mAb (also CAT, UK) which are currently under way, with possible applications in corneal scarring.21
The potential for the use of fully human mAb is great not only in ocular wound healing but throughout the body. Once a target molecule is identified, specific mAbs can be isolated and constructed. Unlike agents such as mitomycin C, which have non-focal and unwanted effects such as inducing cell death, mAbs offer less destructive and focal alternatives with, so far, little evidence of toxicity. However, these neutralising antibodies tend to act at an extracellular level, with inhibitory activity somewhat less than 100% in vivo. Another possible mode of action is to inhibit gene expression of the target molecule.
Modification of gene expression
Although little has been published on the modifying of expression of specific genes involved in scarring in the eye, it has been evaluated elsewhere in the body, for example, as a way of reducing the wound healing response by altering vascular endothelial growth factor (VEGF) expression associated with coronary artery restenosis.22 Since in the eye TGF-β stimulates the scarring response,11 12 19 an obvious strategy is to utilise antisense oligonucleotides and gene transfer techniques with ribozymes to reduce TGF-β gene expression, as discussed in the next three sections.
Antisense oligonucleotides
Antisense oligonucleotides are synthetic molecules that bind to specific intracellular messenger RNA strands (mRNA). They consist of short DNA/RNA sequences, usually of around 20 nucleotides in length, which are designed to be complementary to mRNA strands which code for the production of specific proteins.23-25 By binding to the mRNA molecules, antisense oligonucleotides stop transcription of the mRNA, and hence synthesis of the protein (Fig 3). The exact mechanism by which this occurs is uncertain though triplex formation, blocking RNA splicing, preventing mRNA transport into cytoplasm, increasing RNA degradation, or blocking the initiation of translation have all been suggested.
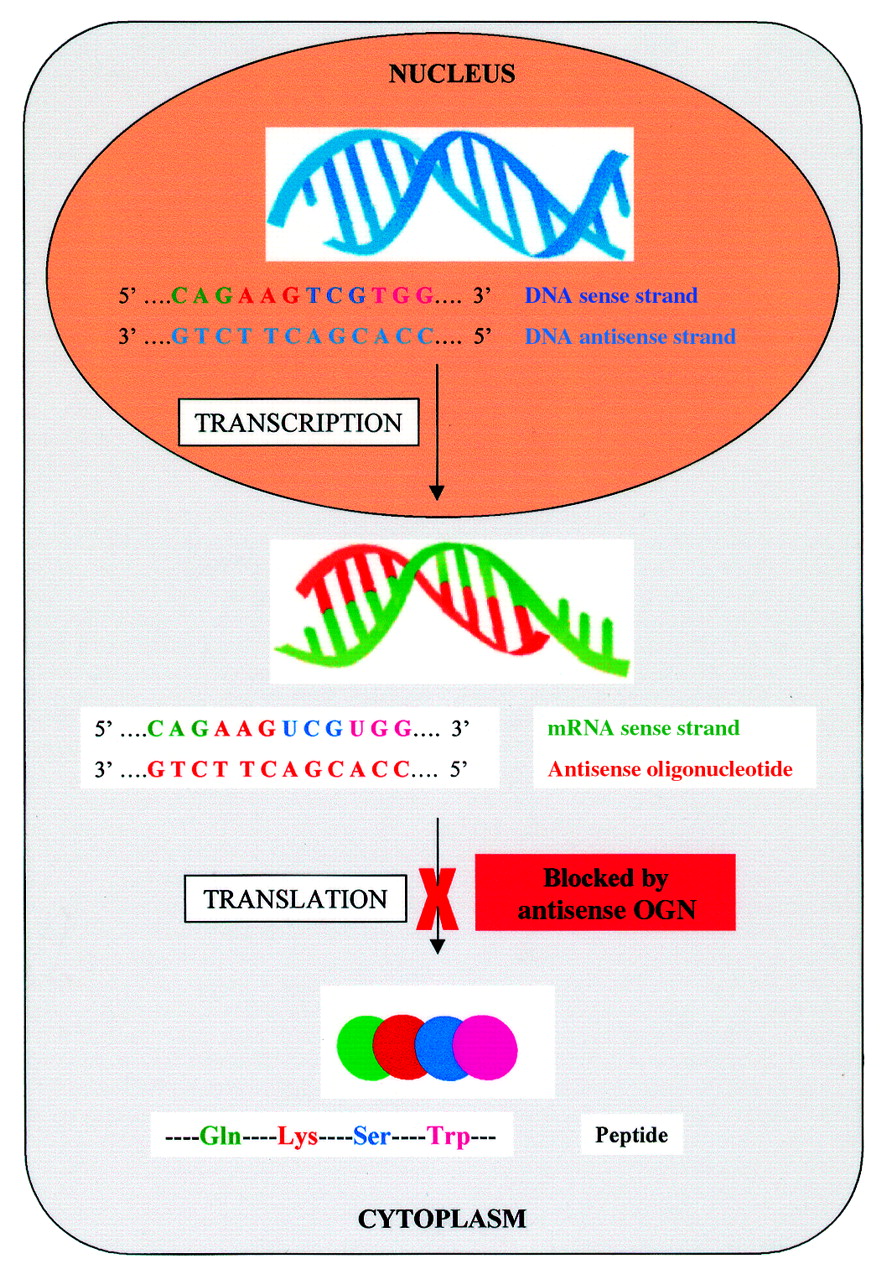
Antisense oligonucleotides. Antisense molecules are synthetic oligonucleotides that have been designed to be complementary to mRNA strands that code for the production of specific proteins. The target mRNA is transcribed from DNA in the nucleus and, on entering the cytoplasm, is bound by the antisense molecule. The binding of the mRNA sense strand stops translation of the mRNA, and hence synthesis of the protein.
Although the concept of antisense therapy is attractive and theoretically simple there are a number of associated problems. Firstly, endogenous cellular nucleases cause rapid cleavage and degradation of antisense molecules. However, modification of the synthetic antisense oligonucleotide phosphodiester bond by replacing an oxygen with sulphur (creating a phosphorothioate backbone) provides resistance to these nucleases and thus increases oligonucleotide stability.26 Secondly, the antisense molecule must have sufficient affinity for targeted mRNA to bind with a high degree of specificity and fidelity. It is essential for the complete sequence of nucleotides of the target mRNA to be known. Theoretically, the minimum number of bases required for an antisense oligonucleotide to bind to a unique DNA sequence is 17 and 13 to an RNA sequence. Modification of the bases in the synthesised oligonucleotides can also increase its affinity to mRNA—for example, the use of 5-methyl C for C.26 Finally, the antisense molecule must be taken up effectively into the cell.
Historically, delivery of antisense oligonucleotides into target cells and/or the cell nucleus has been problematical. The use of various agents such as cationic liposomes and peptides to increase oligonucleotide uptake has met with some success in vitro.27 Recently, there have been promising results with the application of an antisense molecule to the receptor of the growth factor epidermal growth factor, administered enveloped within the cationic liposome lipofectin.28
We are currently investigating TGF-β antisense oligonucleotides and their possible application in reducing conjunctival scarring.
Gene transfer and gene therapy
Gene therapy presents an alternative approach to treating human diseases. Rather than using agents that may only target gene products, gene therapy might be utilised to modify gene expression in a specific manner, and theoretically produce long term effects following a single administration. There are three modes of molecular treatment—gene replacement (where a defective gene is replaced), gene control (where gene expression is modified), and gene addition (expression of a gene not normally found). In wound healing, a possible candidate for gene replacement could be p53 which appears to be defective in keloid formation.29
The process of gene therapy relies on the successful transfer of a gene to specific tissues (Fig 4). This, in turn, is dependent on the vector that is used and the route of administration. A major drawback in the development of effective clinical protocols involving gene therapy has been the disappointing efficiency of gene transfer and expression in patients.30 Compared with other tissues, the eye is an easily accessible target for gene therapy.31 In addition, the possible routes of delivery—for example, subconjunctival injection, topical application, allow localised exposure of the target tissue, with reduced risk of systemic effects. Investigations of in vivo gene therapy strategies involve the use of animal models and we are currently using the mouse and rabbit to evaluate gene therapy vectors in conjunctival and corneal scarring.20 32-34
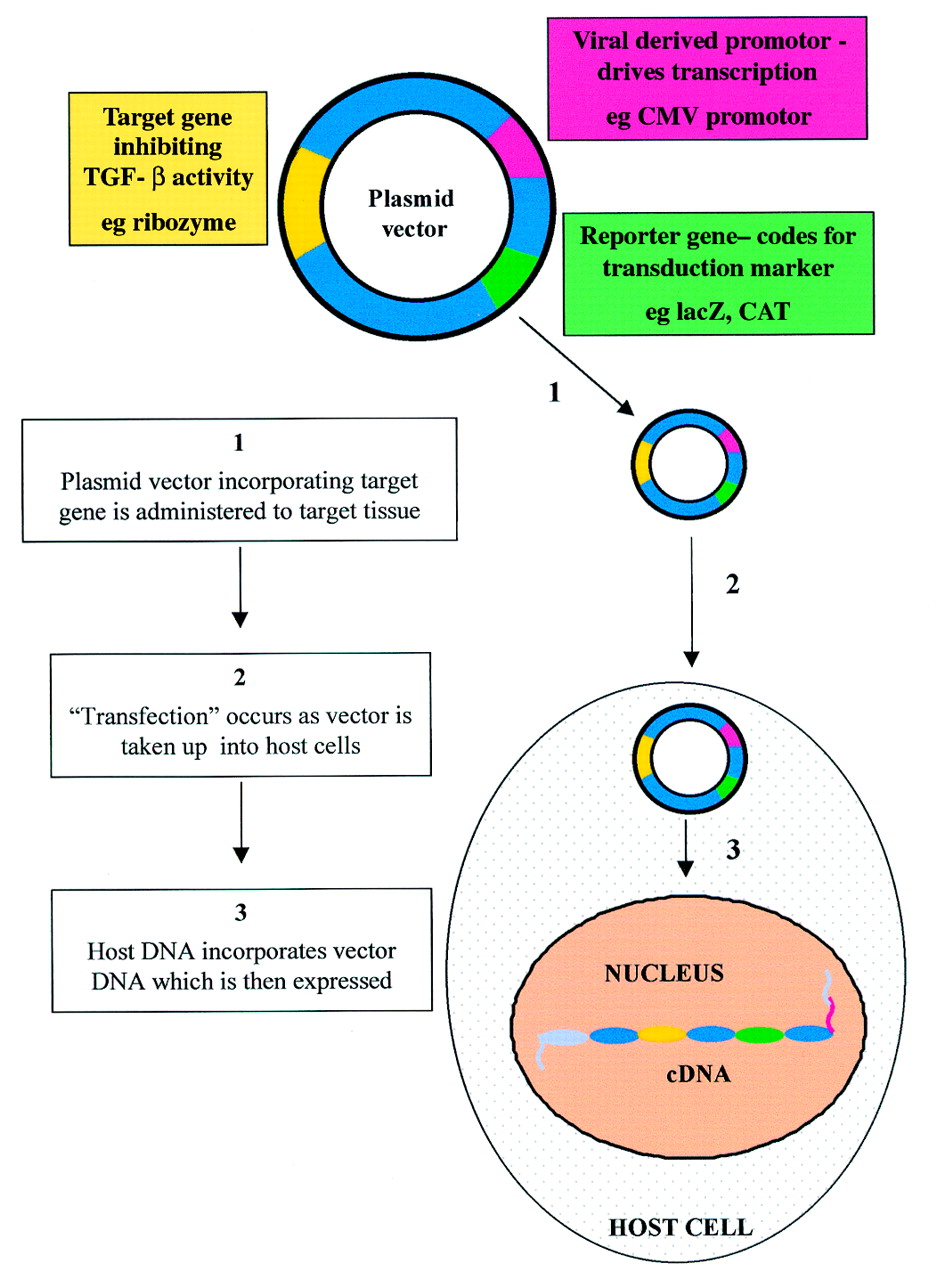
Gene transfer. Gene therapy depends on the successful transfer and delivery of the agents to specific tissues. A DNA vector—for example, a plasmid vector, which includes gene sequences for the target, a viral derived promoter, and a reporter can transfect cells in the target tissue.
There are a number of viral and non-viral vectors currently available. Non-viral gene delivery systems include naked plasmid DNA, ballistic DNA injections, and liposomes. Plasmids are small, autonomously replicating bacterial DNA molecules. They may be designed to express genes in target cells by incorporation of eukaryotic promoters such as the potent cytomegalovirus (CMV) derived promoter sequence (Fig 4). Ballistic DNA injections involve the precipitation of plasmid DNA onto 1–3 μm sized gold or tungsten particles, which are “fired” at the target tissues.35 36 Gene delivery with cationic liposomes is another alternative that has been studied in the eye.37 38 These substances are positively charged liposomes (< 200 nm in diameter) to which bind negatively charged DNA.39 The efficacy of uptake into cells may be further enhanced by addition of peptides which bind to cell surface receptors.40 We are currently studying conjunctival and corneal naked plasmid transduction in vivo models, using reporter genes such as lacZ and chloramphenicol acetyl transferase (CAT). Immunostaining with an antibody against the reporter gene product allows qualitative detection of transduction efficiency and localisation (Fig 5), although quantitative analysis is also possible (for example, CAT ELISA).

Gene transfer in cornea. Using our rabbit model,34 we show transduction of the corneal epithelium with a DNA plasmid expressing a CAT reporter gene has occurred, as detected with Texas red immunostaining (5 μm section, ×20 magnification)
The main disadvantage of the current generation of non-viral vectors is that they only allow short term gene expression generally limited to 3 days. The vector DNA is not stably maintained in the nucleus. In order to achieve longer gene expression it is necessary to use viral vectors. These are viruses which have been modified to contain a marker or therapeutic gene and to be replication deficient. Of the viral vectors, adenoviral (AV) vectors have been the most extensively studied in the eye. Along with other groups, we have demonstrated good AV transduction in ocular tissues. Following intracameral and subconjunctival injections of AV carrying a lacZ reporter gene driven by a CMV promoter, we have demonstrated efficient tranduction of fibroblasts and gene expression for 3 weeks. The bacterial β-galactosidase gene (lacZ) cleaves an artificial substrate (X-gal) to produce a blue reaction product that provides a visible marker of gene expression (Fig6). The major disadvantage of AV vectors is the induction of an inflammatory reaction following administration which results in a loss of transduced cells caused by a cytotoxic immune response.41 Adeno associated virus (AAV) vectors are more attractive than AV vectors, firstly, because the wild type virus is non-pathogenic in humans and, secondly, because the recombinant vectors are less immunogenic. Using AAV it is possible to obtain gene expression for several months. We are currently studying AAV transduction both subconjunctivally and in the cornea.

Gene transfer in conjunctiva. Using our mouse model of conjunctival scarring,33 we have shown that there is transduction of fibroblasts and mononuclear cells in the subconjunctival space with an AV vector expressing a lacZ reporter gene (AV.CMV.LacZnuc). Positive expression is indicated by galactosidase activity seen as blue (nuclear fast red counterstain, 5 μm section, ×80 magnification).
Ribozymes
Ribozymes are RNA molecules that are able to enzymatically cleave specific bonds in other RNA molecules.42-44 Hence, if mRNA of a specific protein is cleaved by a ribozyme, inhibition of protein production occurs (Fig 7). Like antisense oligonucleotides, ribozymes can be designed to target any message, but their significant advantage compared with antisense technology is that the effects of these molecules are not as concentration and time dependent. This is because the ribozyme not only is able to destroy a particular message but also has the capacity to reattach to and destroy other messages of the same sequence many times. In addition, multiple copies of specific mRNA strands are cleaved rapidly at a catalytic rate. Compared with antisense strategies, ribozymes are more stable within cells, forming an enzyme/substrate complex that is relatively resistant to intracellular nucleases.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ribozymes. Ribozymes are RNA molecules that are able enzymatically to cleave specific RNA bonds. Like antisense oligonucleotides, they can be designed to target any mRNA of specific proteins. The cleavage of mRNA by the ribozyme leads to inhibition of translation and protein production.
Several groups have demonstrated reduction of specific mRNA in vitro.45 46 One study has utilised AAV vectors to deliver ribozymes directed against a mutant rhodopsin gene in a rat model of autosomal dominant photoreceptor degeneration.47 48
The future of ribozymes in the management of wound healing in ocular diseases is still to be investigated. Our group has shown that ribozymes directed against TGF-β1 mRNA in vitro successfully reduce expression of TGF-β1 at both protein and mRNA levels within 3 days of application.49 We are currently studying ribozymes to the TGF-β II receptor.
Prospects for molecular therapy
As we move into the new millennium, developments in molecular therapy offer exciting prospects for the modulation of wound healing, not only in the eye but throughout the body. Advances in gene delivery and designs of antisense molecules and ribozymes offer the potential of more specific, safer, focal, and titratable treatment, with far reaching clinical applications.
Acknowledgments
Supported by the Wellcome Trust, UK (grant No 048474) and Medical Research Council.