Article Text

Statistics from Altmetric.com
Cataract is an opacification of the eye lens that frequently results in visual impairment or blindness during infancy and early childhood.1 An estimated 200 000 children are blind with bilateral cataract world wide and 20 000-40 000 children with developmental cataract are born each year.2 In the developed nations of Europe and Northern America, national surveillance or cross sectional studies suggest a prevalence of 1-4 cases per 10 000 children.3–6 In southern India the prevalence is higher, at approximately 6.5 cases per 10 000 children7 and bilateral childhood cataract accounts for about 12% of all ocular disorders registered.8
The lens is a unique tissue as it is separated from the surrounding fluids by the lens capsule and there is life long persistence of its cells and their proteins. The β- and γ-crystallins were biochemically characterised as major lens proteins by Mörner9 over a century ago. They belong to a superfamily of proteins, which were considered for a long time to be present only in the lens. However, it has recently been reported that β- and γ-crystallin mRNA and protein are also present in other tissues, in particular in the retina, brain, and testis.10–12
The common signature of all β- and γ-crystallins is the so called Greek key motif. Crystallography has shown that each of the β- and γ-crystallins is composed of two domains, each built up by two Greek key motifs. It is widely accepted that β/γ-crystallins evolved in two duplication steps from an ancestral gene coding for a protein folded like a Greek key. The γ-crystallin encoding genes (Cryg/CRYG genes) in all mammals consist of three exons: the first one codes only for three amino acids, and the subsequent two are responsible for two Greek key motifs each. Biochemically, the γ-crystallins are characterised as monomers with a molecular mass of 21 kDa.13–15
The family of Cryg genes is mainly located as a cluster of six closely related genes (Cryga→Crygf) on mouse chromosome 1 and on human chromosome 2q33-35; in man, the CRYGE and CRYGF genes are pseudogenes. The seventh Cryg gene (Crygs) is mapped on mouse chromosome 16 and human chromosome 3.14
Several mutations in the Cryg/CRYG genes causing cataracts have been identified in mouse and man. In the mouse, the mutation ENU-436 affects the Cryga gene, the Nop mutation disrupts the Crygb gene,16 the Chl3 mutation destroys the Crygc gene,17 and Lop12 involves the Crygd gene.18 In the murine Cryge gene, four cataract alleles have been reported to date: the Elo mouse,19 the Cat2t mutant,16 the Crygenz mutant,20 and the CrygeAey1 mutant.21 More recently, mutations in the Crygf gene of the mouse have been identified (Graw, Neuhäuser-Klaus, Löster, and Favor, unpublished data). Mutations in CRYG genes have also been implicated in human cataract.22–25 In this study we screened seven Indian families with autosomal dominant congenital cataracts for mutations in the CRYGA-CRYGD genes.
MATERIALS AND METHODS
Clinical documentation
A prospective study on bilateral childhood cataracts (BCCs) was undertaken in collaboration with Aravind Eye Hospital, Madurai, Tamil Nadu (S India) for a period of 10 months (January-October 1995). Patients with bilateral childhood cataracts below the age of 15 years were recruited from the paediatric clinic at Aravind Eye Hospital. The probands and the accompanying parents or relatives underwent clinical eye examination by a senior paediatric ophthalmologist to assess the cataract phenotype through either slit lamp or direct ophthalmoscope depending on the cooperation of the probands. In some unoperated probands and relatives, the phenotype was documented using slit lamp photography. Clinical details were recorded in a standard questionnaire detailing age of onset and diagnosis of cataract as well as general health and parents' medical history including maternal obstetric history. The studies were performed according to the Declaration of Helsinki. All study members received detailed explanation of the study in their regional language before giving informed consent.
Molecular analysis
Since the number of mutations leading to dominant cataracts is fairly high both in human and in mouse Cryg genes, the CRYG gene cluster was tested as a candidate. Therefore, we sequenced the exons and their flanking regions of the four active CRYG genes (A→D) in all probands. Any interesting sequence variation suggestive of a mutation was later confirmed in parents and available relatives by directly sequencing the particular exon of the respective CRYG gene. The designations of such variations and mutations follow the recommendations of a standard nomenclature system for human gene mutations.26
In total, during this study 12 subjects from seven cataract families and one control family were investigated. Additional members of both affected and unaffected status were screened whenever required to confirm any sequence variation or polymorphism. All samples came from subjects of Indian origin (except one from an unaffected person from Europe). A total of 5-10 ml of venous blood was collected from the available affected and unaffected members of the families. Genomic DNA was isolated as previously described.27
PCR was performed on genomic DNA samples in reactions of 20 μl with a denaturation step at 95°C for two minutes for one cycle, followed by 40 cycles of 95°C denaturation for 45 seconds, annealing at 60°C for 45 seconds, extension at 72°C for 45 seconds, and with a final extension at 72°C for five minutes using either a Stratagene Robocycler (Stratagene, Heidelberg, Germany) or a Perkin Elmer Thermocyler (Perkin Elmer, Weiterstadt, Germany). The primers are listed in table 1.
Oligonucleotides used as primers for PCR amplification of human CRYG genes
Sequence analysis was performed commercially (SequiServe, Vaterstetten, Germany) after purification of PCR fragments through Nucleospin™ extraction columns (Macherey-Nagel, Düren, Germany). Computer assisted prediction of the biochemical properties of the changed proteins used the Proteomics tools of the ExPASy server (http://www.expasy.ch). Putative functions of changes in the γ-crystallin promoters were illuminated by the software package of MatInspector professional (http://genomatix.gsf.de28).
RESULTS
Seven families with autosomal dominant childhood cataracts were identified from the hospital database during the study period of January to October 1995 at the Aravind Eye Hospital, Madurai, India.
Polymorphic sites in the CRYG genes
A variety of sequence variations depicting single nucleotide polymorphisms was observed in all CRYG genes of the cataract probands and control subjects of Indian origin in coding as well as in non-coding intronic regions as compared to the GenBank/EMBL database (table 2). Some of these sequence changes in coding regions even lead to alterations of amino acids, for example, L148P (CRYGA, exon 3), S73I (CRYGB, exon 2), L111I (CRYGB, exon 3), R48H (CRYGC, exon 2), and V101M (CRYGD, exon 3). Of these, L148P and V101M appear to be the only allele occurring in this population, while R48H and S73I appear to be quite infrequent. Moreover, two other allelic forms were found to be different from the database entries in all subjects analysed (one silent polymorphism, one in a non-coding region). All these alleles were homozygous.
Polymorphisms in human CRYGA-CRYGD genes in subjects of Indian origin
Identification of mutations in CRYG genes associated with human cataracts
In three of the seven probands, unique mutations in one of the four CRYG genes could be identified as cosegregating in a heterozygous condition with the disease in the respective family.
Family C135
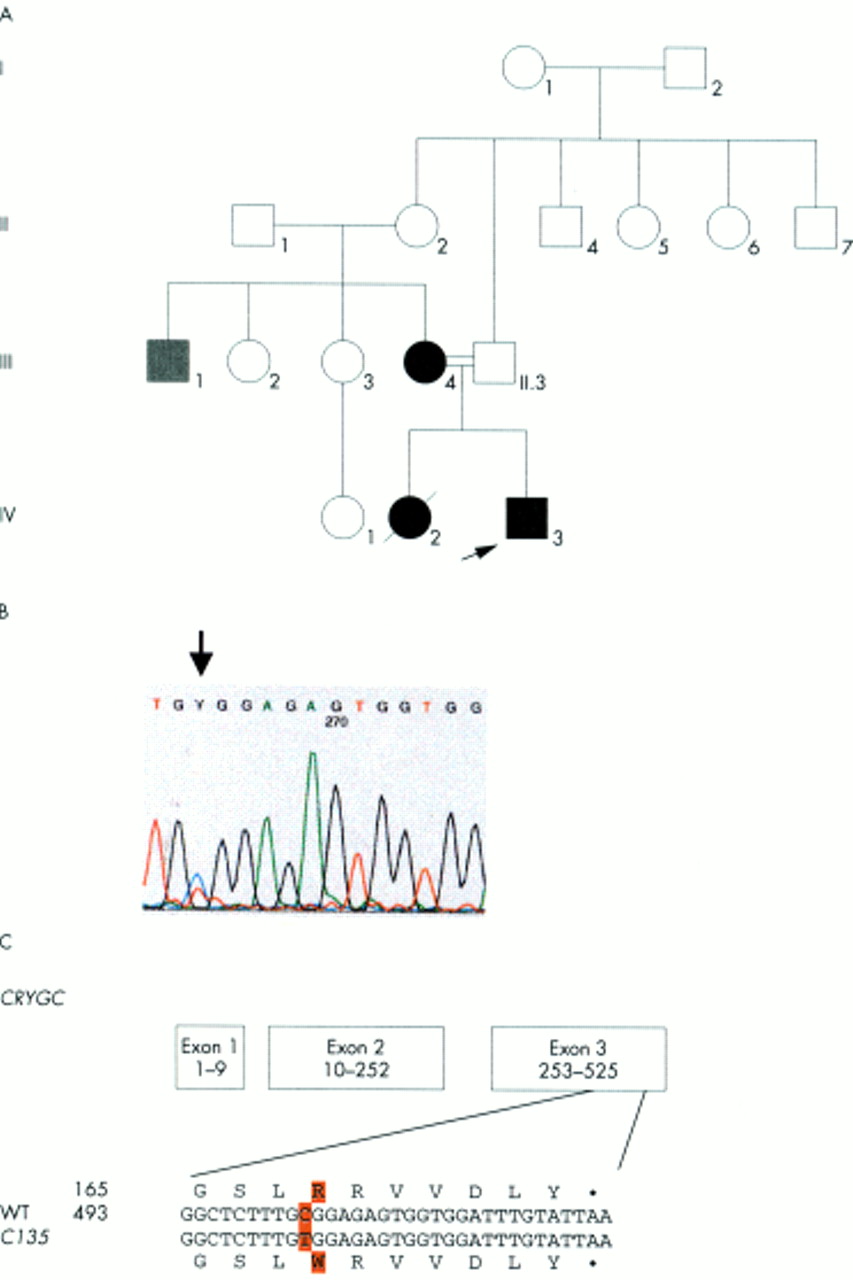
In family C135 (fig 1A), the congenital lamellar cataract is characterised by a mutation in CRYGC: a point mutation in exon 3 (502C→T) leads to a replacement of an Arg at position 168 by Trp in the fourth Greek key motif (fig 1B, C). The Arg at this position is highly conserved and present in all γ-crystallins of mouse, rat, ox, and man. The mutation cosegregates with the disease in the family (IV.3, III.4); the mutation was not observed in unaffected family members (III.3, II.1) nor in control subjects of Indian descent. The maternal uncle (III.1) has a different clinical entity and also does not show this mutation.
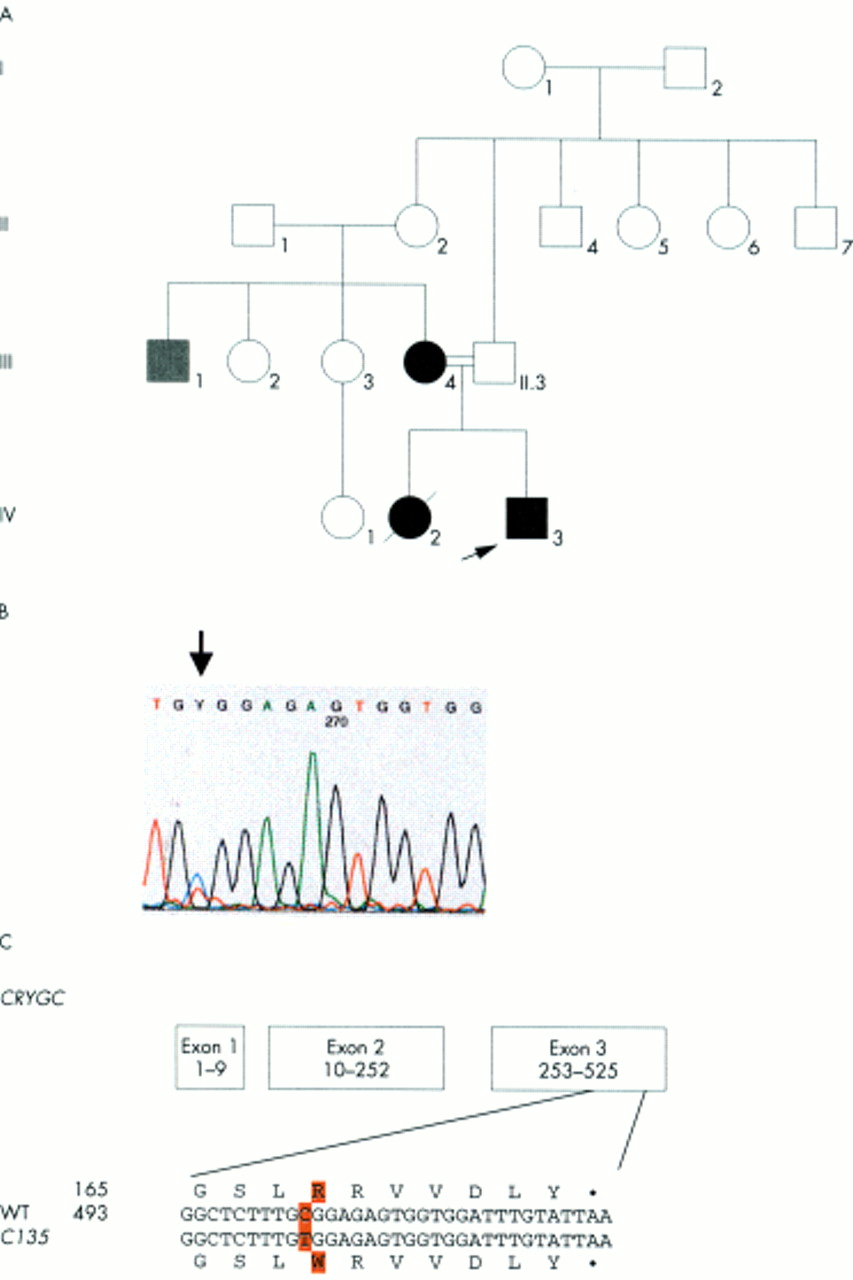
Analysis of family C135. (A) The pedigree of family C135 indicates the autosomal dominant inheritance of the congenital lamellar cataract. The proband is marked by an arrow. (B) Exon 3 of the CRYGC gene was amplified by PCR. Sequence analysis of the amplified product showed a heterozygous position of both C and T, resulting in Y of the sequence chromatogram (indicated by an arrow). (C) The C→T exchange at position 502 leads to an amino acid alteration from Arg to Trp at position 168. The mutation was observed in heterozygous condition in the affected members of the family (III.4, IV.3), but not in the others screened (II.1, II.3). III.1 had cataract, but was also microcephalic and mentally retarded.
Family C87
In family C87, congenital lamellar cataract (fig 2A) was observed in the proband and her affected father (fig 2B). The phenotype cosegregates with the point mutation in exon 2 of CRYGD (70C→A) that leads to the replacement of a Pro by a Thr at position 23 (fig 2C, D). At this particular position, the Pro is present in all human γ-crystallins as well as in rat and mouse γA-, γB-, and γC-crystallins. This mutation was observed in both affected family members (II.2, I.2). The mutation is unique among all the samples analysed in this study; therefore, it may well be considered as the molecular basis of the phenotype.

Analysis of family C87. (A) The clinical picture of the proband aged 3 years shows bilateral congenital lamellar cataract. (B) The pedigree of family C87 indicates the autosomal dominant inheritance of the congenital lamellar cataract. The proband is marked by an arrow. (C) Exons 1 and 2 of the CRYGD gene were amplified by PCR. Sequence analysis of the amplified product showed a heterozygous position of both C and A, resulting in M of the sequence chromatogram (indicated by an arrow). (D) The C→A exchange at position 70 leads to an amino acid alteration from Pro to Thr at position 23. The mutation was observed in heterozygous condition in the proband (II.2) and her affected father (I.2). The mutation was not observed in other probands and the control of Indian descent.
Family RCS91
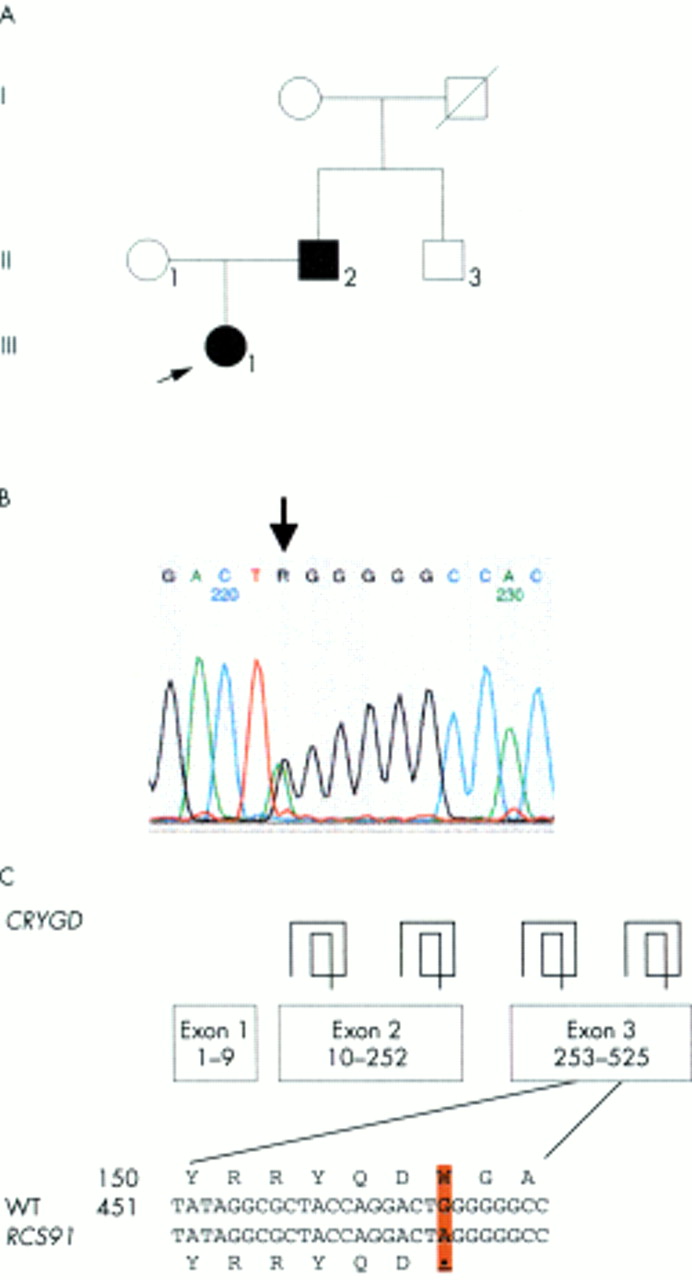
The proband of family RCS91 (fig 3A) suffered from a congenital central nuclear cataract. The cataract is most likely caused by a point mutation in the third exon of CRYGD (470G→A) leading to a premature stop after 156 amino acids (fig 3B, C) thus resulting in a truncated protein missing 18 amino acids at the C-terminus. The fourth β sheet of the fourth Greek key motif might not be formed leading to a major change in structural conformation. This mutation was observed only in the proband (III.1) and her affected father (II.2); the unaffected mother did not show this mutation. Also, such a change was not observed in the other cataract probands nor in the unrelated controls representing the unaffected population of the same ethnic group.
{kind=link}
{kind=link}
{kind=link}
Analysis of family RCS91. (A) The pedigree of family RCS91 indicates autosomal dominant inheritance of the congenital central nuclear cataract. The proband is marked by an arrow. (B) Exon 3 of the CRYGD gene was amplified by PCR. Sequence analysis of the amplified product showed a heterozygous position of both G and A, resulting in R of the sequence chromatogram (indicated by an arrow). (C) The G→A exchange at position 470 leads to a premature stop codon at codon 156. The mutation was observed in heterozygous condition in the proband (III.1) and her affected father (II.2), but not in the unaffected mother (II.1). The same mutation was not observed in other probands with cataracts or the control of Indian descent screened in this study.
Four additional families
In four additional families, no mutations in the CRYG genes were observed. The phenotypes were nuclear since birth in all affected members of families C99, C107, and C176, while lamellar cataract is present in family C132. Proband C99 of consanguineous origin had zonular cataract with nuclear opacity and microcornea like the affected father and a sib. Proband C107 was 3 years old at the time of registration with an affected father having nuclear cataract since birth. Proband C132 was 4 years old and had lamellar cataract. It was stationary and non-progressive in all affected members. Proband C176 was 6 years old with central dense nuclear cataract and affected members spread out in three generations. Since no mutations cosegregated with the CRYG genes, these probands need to be investigated further for the causative mutations.
DISCUSSION
In this study we have identified the causative mutations in three families with autosomal dominant congenital cataracts. Two families showed a lamellar cataract phenotype and one a central nuclear cataract. All three cataract phenotypes are caused by mutations in exons 2 or 3 of either the CRYGC or CRYGD genes.
An increasing number of mutations in the CRYG genes have been described in association with human congenital cataract. Coppock-like cataract is caused by a mutation in the CRYGC gene and aculeiform cataract and a progressive punctate cataract with mutations of CRYGD.22,23 Furthermore, a 5 bp insertion in the γC-crystallin gene (CRYGC) has been shown to be associated with a dominant, variable zonular pulverulent cataract.25 Quite recently, biophysical characterisation of a novel CRYGD allele (C109A) involving a substitution of an Arg residue at codon 36 of the γD-crystallin by a Cys has been shown to lead to crystallisation of the protein in the lens.24 All mutations characterised so far in the human CRYG genes are listed in table 3. A polymorphic congenital cataract has also been mapped very close to the CRYGB gene.29 It is noteworthy that all known mutations were found only in the CRYGC and CRYGD genes, but none in CRYGA or CRYGB. This may be because these are the most expressed members of the CRYG gene family in man.30 Interestingly, the two different mutations characterised in two lamellar phenotypes in our study further strongly support the genetic heterogeneity of congenital cataracts.31,32 Obviously, it is impossible to deduce the underlying molecular lesion from the clinical observation or, alternatively, to predict the resulting phenotype from the characterised mutation.
Human CRYG mutations in autosomal dominant congenital cataract
In mice, the number of mutations characterised in the Cryg genes is also extensive; Crygeelo19 and CrygdLop1218 have been reported by other groups, while we have characterised seven murine cataract mutants, Cryga1Neu, Crygbnop, CrygcChl3, Cryget, Crygens, Crygenz, and CrygeAey1.16,17,20,21,33 Among them, the CrygdLop12 mutant18 is identical (470G→A, W156X) to the sequence change observed in our pedigree RCS91. It is predicted18 that this mutant protein might have altered protein folding of the γ-crystallins and thus result in lens opacity. However, there is only a partial overlap of the phenotypes; the opacity was characterised in family RCS91 as a central nuclear cataract, and in the Lop12 mouse as a dense, irregular nuclear cataract with mild cortical opacification at a later age. Such phenotypic variations between two species have been described previously.1
There are several reports both in mice and in humans of polymorphic sites within these genes without apparent effects on the function of the proteins.18,22 In particular, the change L148P in the CRYGA gene was present in all subjects investigated during our study. The Pro residue at this position appears to be highly conserved in all other γ-crystallins of man, mouse, rat, and ox. Therefore, the given sequence in the GenBank/EMBL database (Acc No M17315) needs to be ascertained in diverse ethnic groups.
Another allele of very high frequency is V101M in γD-crystallin. The Val residue at this position of this crystallin is reported in both mouse and human, and also in the γA-crystallin of mouse and rat at a comparable position. In other crystallins, either Met or Ile residues are present at this position. All subjects analysed in our study showed this 304G→A (V101M) allele in the homozygous state, which is in agreement with two GenBank/EMBL database entries (XM_002461 and HSU66583); the sequence K03006 should be considered as an exception.
Three other polymorphic sites are of interest: first, the change 198G→A (Acc No M17315) of intron A in CRYGA seems to occur at a fairly high frequency. It remains to be elaborated whether such a change at a position 20 bp upstream of the 3` end of intron A might influence the ensuing splicing mechanism.
Secondly, the allele –47T→C affects the promoter of the CRYGB gene and occurs in five out of 10 cases in a heterozygous condition. This nucleotide substitution destroys a putative Ikaros1 binding site, which is located between the TATA box and the transcription initiation site. Ikaros proteins consist of unique combinations of zinc finger modules34 featuring them as interesting transcription factors. However, it remains to be investigated whether Ikaros gene(s) are actively transcribed in the lens and if they bind to any of the CRYG promoters.
The third interesting polymorphic site destroys the polyadenylation signal in the CRYGD gene (564A→T). This mutation was observed in two subjects and might influence the stability of the CRYGD transcript. However, the loss of a CRYG transcript may not necessarily lead to cataract formation as shown by the evolutionary conversion of the Cryge and Crygf genes to pseudogenes in man.35 It is unlikely, therefore, that this particular mutation is the cause of the cataract formation in family C132.
-
γ-crystallins are crucial in the maintenance of lens transparency and in mammals they are expressed mainly in the ocular lens.
-
Seven families with non-syndromic, autosomal dominant congenital cataracts were screened for mutations in genes encoding γ-crystallins (CRYG).
-
DNA was extracted from blood samples provided by the probands, their parents, and other available family members. The coding and flanking regions of γA-γD-crystallin encoding genes (CRYGA-CRYGD) were amplified by PCR from genomic DNA and sequenced.
-
In three families, two missense mutations and one nonsense mutation in the coding regions of a CRYG gene were found to cosegregate with the phenotype. The mutations were characterised as R168W in CRYGC of family C135, P23T in CRYGD of family C87, both of which had a lamellar cataract phenotype, and W156X in CRYGD of family RCS91 with central nuclear cataract. The latter mutation is identical to the dominant Lop12 cataract in mouse. Several interesting polymorphic sites in the CRYG genes are documented in subjects of Indian origin.
-
This study further confirms the CRYG gene cluster as a major locus in cataractogenesis in humans. The identification of similar cataract causing mutations in mice and humans will facilitate further work to elucidate the mechanisms of lens opacification associated with mutations of CRYG genes and how the effects of the primary mutation may be influenced by a varying genetic background.
Future γ-crystallin research should critically analyse the significance of such a large number of polymorphic sites in affected families of Indian origin and whether they may be responsible for recessive forms of cataracts. A probe for such polymorphic sites in the affected population may explain the genetic susceptibility and the underlying genomic diversity in different ethnic groups. Moreover, the finding of autosomal dominant congenital cataracts being associated with mutations in other Cryg genes further implicates the CRYG gene cluster as a very critical locus for lens development and differentiation. The occurrence of several diverse clinical phenotypes further supports the vital role of these genes in conferring lens transparency.
Acknowledgments
The expert technical assistance of Erika Bürkle, Dagmar Reinl, and Monika Stadler is gratefully acknowledged. Oligonucleotides were obtained from Utz Linzner (GSF-Bioinformatics Group, Institute of Experimental Genetics). The authors also thank Dr A T Moore (London/Cambridge) for critical reading of the manuscript. STS and JG acknowledge financial support from the DLR, Bonn, Germany, and ICMR, Government of India, New Delhi for official clearance of the project (No IND 99/021) and the families for their kind cooperation.