Article Text

Abstract
Uveal melanoma represents ∼85% of all ocular melanomas and up to 50% of patients develop metastatic disease. Metastases are most frequently localised to the liver and, as few patients are candidates for potentially curative surgery, this is associated with a poor prognosis. There is currently little published evidence for the optimal management and treatment of metastatic uveal melanoma and the lack of effective therapies in this setting has led to the widespread use of systemic treatments for patients with cutaneous melanoma. Uveal and cutaneous melanomas are intrinsically different diseases and so dedicated management strategies and therapies for uveal melanoma are much needed. This review explores the biology of uveal melanoma and how this relates to ongoing trials of targeted therapies in the metastatic disease setting. In addition, we consider the options to optimise patient management and care.
- Choroid
- Ciliary body
- Iris
- Drugs
- Treatment Medical
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Biology of uveal melanoma
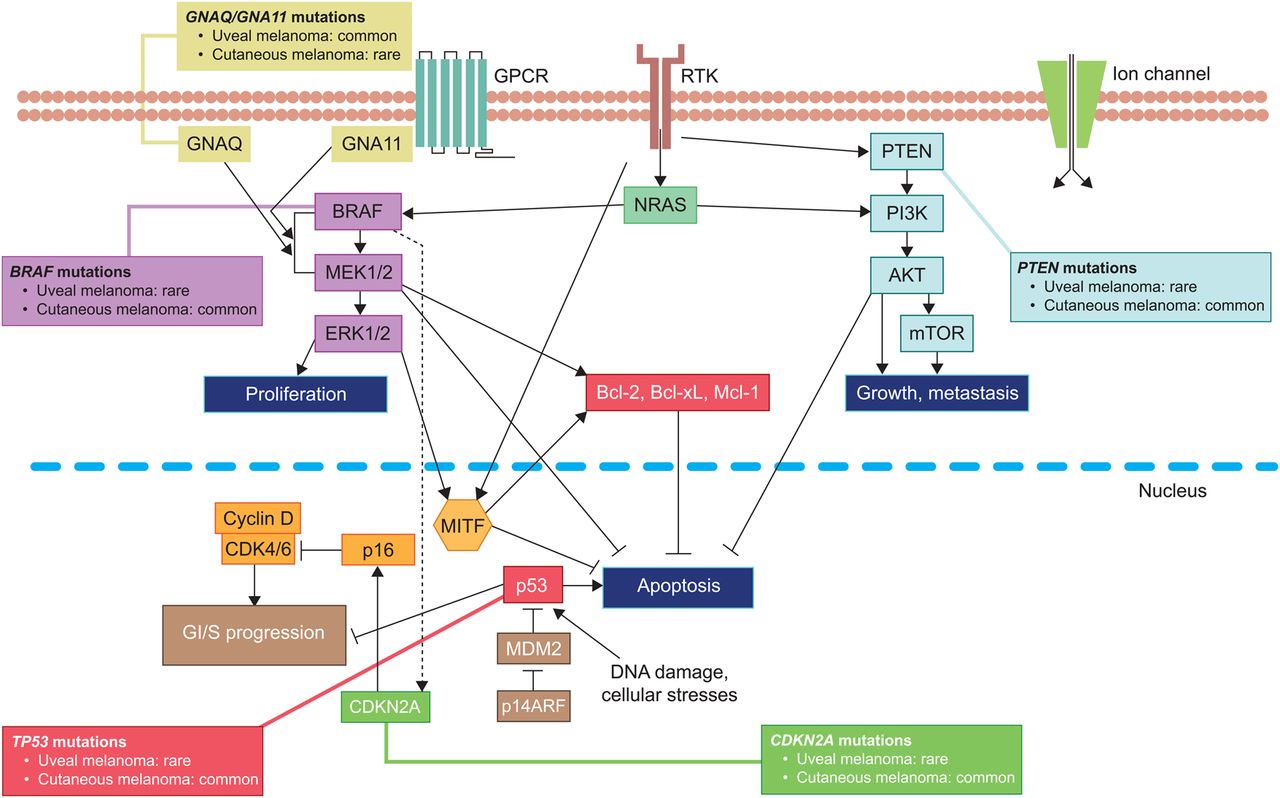
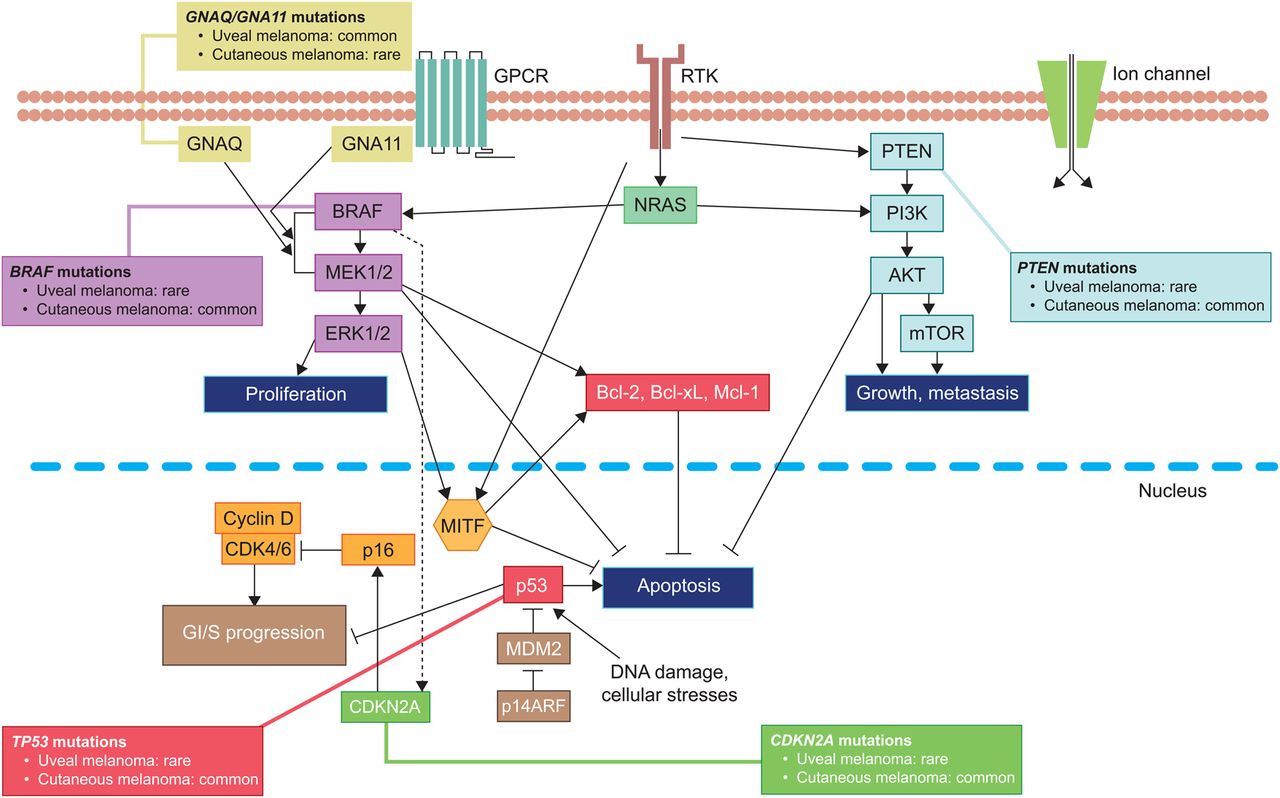
Uveal melanoma is the most common primary intraocular malignancy in adults, representing ∼85% of ocular melanomas.1 Remaining ocular melanomas arise from the conjunctiva (∼5%) or other sites (∼10%).1 Uveal melanoma is considered a rare cancer, representing ∼3%–5% of recorded melanoma cases in the USA.1 ,2 Unlike cutaneous melanoma, the most common subtype, which arises from melanocytes located in the basal layer of the epidermis,3 uveal melanoma arises from melanocytes located anywhere in the uveal tract. Approximately 85%–90% of cases involve the choroid, while those remaining are localised to the iris or ciliary body.4 ,5 Cutaneous and uveal melanomas are biologically distinct (figure 1) and differ in terms of incidence by gender, race and geographical area.4
Incidence
By contrast to rates of cutaneous melanoma, which have been steadily increasing since the 1970s,6 the incidence of uveal melanoma has remained stable for many years.2 ,7 In the USA, the mean age-adjusted incidence is 5.1 per million,2 while incidence in Europe varies with latitude being greater in Northern (≥8 cases per million in Norway and Denmark) compared with Southern (two cases per million in Spain and Italy) Europe.7 Risk factors for the development of uveal melanoma include Caucasian ethnicity, welding, light eye colour (green or blue), dysplastic naevus syndrome, ocular melanocytosis and presence of germline BRCA1-associated protein 1 (BAP1) mutations.8–13
Epidemiology/tumour biology
The molecular profile of uveal melanoma is different from those of cutaneous or mucosal melanomas and is composed of a number of chromosomal abnormalities and somatic gene alterations (figure 1).
Monosomy 3, 1p loss, 1q gain, 6q loss, 6p gain, 8p loss and 8q gain are common chromosomal abnormalities in uveal melanoma.14 ,15 Ten-year disease-specific mortality rate varies with abnormality.14 ,15 Monosomy 3 is observed in ∼50% of tumours and is associated with metastatic disease.16 Simultaneous monosomy 3 and chromosome 8 alterations are associated with a worse prognosis, while the outcome is more promising in patients with tumours harbouring partial monosomy 3.16 ,17
Oncogenic mutations in genes associated with the G-protein-α subunits GNAQ or GNA11 are observed in ≥80% of primary uveal melanomas and are associated with constitutive activation of signalling pathways including the central oncogenic RAS/RAF/MEK/ERK (RAS-ERK) pathway;18–20 thereby driving cell proliferation, tumour growth and progression21 ,22 (figure 1).
Inactivating BAP1 mutations are found in ∼50% of all cases, most frequently in class 2 metastasising disease.13 ,23 BAP1 encodes the catalytic subunit of a nuclear ubiquitin carboxy-terminal hydrolase, with various substrates including BRCA1 and histone H2A.24 ,25 Inactivating BAP1 mutations increase the prometastatic behaviour of uveal melanoma cells, although the mechanism by which this occurs remains unknown.24
Mutations associated with a less aggressive behaviour include those found in splicing factor 3B subunit 1 (SF3B1) and eukaryotic translation initiation factor 1A, X-linked (EIF1AX).26 ,27 These mutations are mutually exclusive to one another in 19% and 24% of uveal melanomas, respectively.26 ,27 Despite this frequency, SF3B1 or EIF1AX mutations are found in just 3% of uveal melanomas with monosomy 3.27 Furthermore, preliminary whole genome single-nucleotide polymorphism microarray data showed that amplification of CNKSR3, the product of which is thought to be involved in transepithelial sodium transport, correlated with improved survival of patients with primary uveal melanoma.28
Prognostication
Survival prognostication takes into account clinical predictors (basal tumour diameter, tumour thickness, ciliary body involvement, extraocular spread), pathological predictors (epithelioid melanoma cytomorphology, presence of extravascular matrix patterns, high mitotic count per 40 high-power fields) and genetic predictors, as described above.29 Most histopathological indicators show good correlation with metastatic mortality and mathematical methods have been developed to combine the clinical tumour, node, metastasis stage30 with pathological and genetic prognostic factors. Personalised prognostication is a matter of debate between physicians as many believe that there is no advantage in predicting an unpreventable fatal outcome. However, studies have shown that patients find an uncertain prognosis more stressful than a poor one and accurate prognostication allows special care to be targeted at high-risk patients.29
Metastatic disease
Despite the development of effective local therapies, 5-year survival rates (∼80%) have not changed in the past three decades2 and up to 50% of patients develop metastases.31 No effective adjuvant systemic therapy has been demonstrated to reduce the risk of metastasis, as recently reviewed by Triozzi and Singh.32 One-year survival of patients with metastases is reported to be 15%, with reported median survival ranging from 4 to 15 months.33–35
Surveillance to identify high-risk patients
Given the poor prognosis associated with the development of metastatic disease, early identification of patients, following the treatment of primary disease, who are at high risk of metastasis may be of value and could allow for tailored management of patients. The risk of developing metastatic disease is determined by multiple factors, including tumour size, location36 and genetic profile. Gene expression profiling tests are one way to determine a patient's risk of progressing; patients can be classified as class 1 (low metastatic risk) or class 2 (high metastatic risk),37 with approximately 40% of patients falling into the latter group. Despite being at lower risk, as classified by genomic profiling, approximately 15% of patients with uveal melanoma who experience metastatic disease are class 1; these patients comprise a distinct molecular subgroup to those class 1 patients whose disease does not metastasise, including a striking increase in expression of preferentially expressed antigen in melanoma.38 Genotypic profiling using multiplex ligation-dependent probe amplification has also proven useful for determining high-risk patients.14 About 65% of patients defined as high risk relapse within 5 years,39 suggesting that surveillance of at least this duration should be undertaken.40
Treatment options for metastatic disease
For those patients who do develop metastatic disease, there is as yet no proven standard of care. Dacarbazine, a chemotherapeutic option for treatment of cutaneous melanoma, has been used for uveal melanoma, despite the inherent differences between these molecularly distinct diseases,40–42 and activity has been limited.41 ,43 ,44 Other chemotherapeutic regimens including temozolomide, cisplatin, treosulfan, fotemustine and various combinations have been investigated in uveal melanoma with disappointing results to date.34 ,45 ,46
Ipilimumab, a human monoclonal antibody that blocks the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), is approved in the USA and Europe for the treatment of advanced, unresectable melanoma.41 Response rates of ∼5%–10% have been reported from evaluations in metastatic uveal melanoma,47–50 but evidence of a median overall survival (OS) of 6.0–9.7 months in these trials suggests that responses could be delayed and durable in only a minority of patients.48 ,49 Preliminary data from the phase II GEM-1 trial in treatment-naïve patients suggested more promising response rates than previously reported.51 However, ipilimumab demonstrated very limited clinical activity in treatment-naïve or pre-treated patients with metastatic uveal melanoma in the phase II DeCOG trial; median progression-free survival (PFS) was 2.8 months and median OS was 6.8 months.52
Nivolumab and pembrolizumab, fully human monoclonal antibodies targeting the programmed cell death 1 (PD-1) receptor are approved in the USA and Europe for advanced melanoma.53–55 However, the activity of PD-1 inhibition in uveal melanoma has not yet been well described. Initial assessment of pembrolizumab in seven patients with metastatic uveal melanoma who had progressed on ipilimumab reported a median PFS of ∼3 months;56 a phase II trial in patients with metastatic uveal melanoma is currently recruiting (NCT02359851).
There is a real need for specifically approved treatments and dedicated management strategies in order to improve outcomes for patients affected by this difficult-to-manage disease. Given the limited activity of currently approved agents for advanced melanoma in the treatment of metastatic uveal melanoma, efforts have been placed on conducting clinical trials that have been designed based on our increased understanding of the biology of this disease.
MEK inhibitor trials in the metastatic setting
GNAQ/GNA11 mutations drive the constitutive activation of the RAS-ERK pathway. However, uveal melanomas lack the oncogenic aberrations of this pathway such as BRAF,57 which mediate sensitivity to the BRAF inhibitors vemurafenib and dabrafenib in cutaneous melanoma.58 ,59 Given the molecular profile of uveal melanoma, there is a rationale for treatments that target downstream components of the molecular pathways driving tumour growth, including MEK and protein kinase C (PKC).
Reduction of uveal melanoma cell viability with selumetinib (AZD6244, ARRY-142886), an oral, potent and highly selective, allosteric MEK1/2 inhibitor60 with a short half-life,61 ,62 was correlated with a MEK-dependent gene signature and found to be greater in tumour cells with GNAQ or BRAF mutations than in wild-type cells.63
In a phase II trial in 101 treatment-naïve or pre-treated patients with metastatic uveal melanoma, treatment with selumetinib resulted in improvements in efficacy outcomes compared with chemotherapy (temozolomide or dacarbazine).43 Median PFS was significantly improved with selumetinib (15.9 vs 7 weeks; HR, 0.46 (95% CI 0.30 to 0.71); one-sided p<0.001).The corresponding response rate was 14% with selumetinib compared with 0% with chemotherapy. No significant improvement in median OS was observed (11.8 vs 9.1 months for selumetinib and chemotherapy, respectively (HR, 0.66; 95% CI 0.41 to 1.06)).However, assessment of OS was confounded by the crossover of 86% of patients from the chemotherapy arm to selumetinib, as it was observed that prior treatment with temozolomide or dacarbazine may affect the efficacy of selumetinib.43
Based on these promising observations, a phase III trial (n=129) to assess the efficacy of selumetinib in combination with dacarbazine in patients with systemic treatment-naïve metastatic uveal melanoma was initiated (SUMIT, NCT01974752).64 This was the first clinical trial in uveal melanoma designed with intent to register a drug product with regulatory bodies. However, SUMIT did not meet its primary end point of PFS by blinded independent central review (BICR).65 Median PFS was not significantly improved in the selumetinib+dacarbazine arm compared with the placebo+dacarbazine arm (2.8 vs 1.8 months; HR 0.78 (95% CI 0.48 to 1.27); two-sided p=0.32); the corresponding response rates were 3.1% and 0%, respectively (two-sided p=0.36). There was a numerical improvement in investigator-determined median PFS in the selumetinib+dacarbazine arm (3.8 vs 2.1 months; HR 0.49 (95% CI 0.28 to 0.84)). Further evaluation of the discrepancies between BICR and investigator-determined PFS and assessment of biomarkers are ongoing. OS data were immature at the time of primary analysis and no further analyses are foreseen based on trial assumptions.
Differences in patient population, study design and notably the addition of dacarbazine to selumetinib in SUMIT, compared with selumetinib monotherapy in the phase II trial43 may have led to the differences observed in PFS between the two trials. Further assessment of selumetinib for the treatment of uveal melanoma is ongoing in a trial comparing weekly intravenous paclitaxel 80 mg/m2 in combination with selumetinib 75 mg and weekly intravenous paclitaxel 80 mg/m2 in combination with selumetinib 75 mg twice daily with 2 days off prior to each paclitaxel bolus (EudraCT: 2014-004437-22). A second phase I trial is being developed in which selumetinib will be escalated above 75 mg twice daily using an intermittent dosing schedule of 3 days on followed by 4 days off.
Trametinib, another potent MEK1/2 inhibitor, is characterised by different pharmacokinetic properties than selumetinib, including a longer half-life.61 ,66 In a phase I trial in advanced melanoma, trametinib demonstrated limited clinical activity in 16 heavily pre-treated patients with metastatic uveal melanoma, corresponding to a median PFS of 1.8 months and an unconfirmed overall response rate of 0%.67 No radiographic responses were observed; two patients achieved 24% tumour reduction and 8 achieved stable disease. Four patients received treatment for ≥16 weeks; two patients received treatment for ≥40 weeks.67
Phosphorylated AKT is observed in >50% of uveal melanomas and is associated with a higher risk of metastatic disease.68 A phase II trial prospectively evaluating the efficacy of trametinib with or without the AKT inhibitor GSK2141795 in patients with metastatic uveal melanoma is ongoing (NCT01979523; table 1). As selumetinib in combination with the AKT inhibitor MK2206 reduced cell viability by >50% in multiple GNAQ-mutant uveal melanoma cell lines and reduced tumour volume in mouse xenograft models,69 the combination of MEK and AKT inhibition is a treatment strategy of interest.
Ongoing clinical trials in metastatic uveal melanoma
Activating somatic mutations in GNAQ led to the constitutive activation of the downstream PKC pathway PLCβ/PKC/ERK1/2.70 Preclinical in vitro models showed a strong sensitivity to synergistic treatment with the MEK inhibitor binimetinib (MEK162) and the PKC inhibitor AEB071 (sotrastaurin), associated with halting of proliferation and induction of apoptosis. The combination also significantly reduced tumour size in a GNAQ-mutant mouse xenograft model.71 The combination of binimetinib and AEB071 is being investigated in a phase Ib/II trial in patients with metastatic uveal melanoma (NCT01801358).
Other trials in the metastatic setting
The multi-targeted tyrosine kinase inhibitor, sunitinib, inhibits driver mutations in the receptor tyrosine kinase, c-Kit. Immunohistochemistry analysis found that c-Kit was expressed in ≥63% of surgical primary uveal melanoma specimens, suggesting that c-Kit may be important in uveal melanoma tumour growth.72 Median PFS (2.76 vs 3.88 months (HR, 1.09; 95% CI 0.62 to 1.92)) and OS (6.35 vs 8.65 months (HR, 1.59; 95% CI 0.86 to 2.96)) were not improved with sunitinib over dacarbazine in a phase II trial of 74 patients with metastatic disease who had received no prior systemic therapy for advanced disease.44
Bevacizumab is an antiangiogenic agent that targets all isoforms of vascular endothelial growth factor (VEGF); overexpression of VEGF in uveal melanoma results in increased tumour size and angiogenesis.73 ,74 In preclinical models, bevacizumab inhibited VEGF activity, reducing primary ocular melanoma growth and suppressing the formation of hepatic micrometastases.75 In a retrospective review of uveal melanoma treatments, no significant survival benefit was seen in patients treated with bevacizumab.76 No objective responses were observed in a phase II trial of the combination of bevacizumab and temozolomide (n=35); median PFS and OS were 3 and 12 months, respectively.77
Activating mutations of GNAQ and GNA11 upregulate the hepatocyte growth factor, MET, which is implicated in the metastasis of uveal melanoma.78 Clinical activity in patients with uveal melanoma has been achieved with cabozantinib, a non-selective dual MET and VEGF inhibitor.79 Subset analysis of 23 patients with uveal melanoma treated as part of a phase II trial revealed a median PFS and OS of 4.8 and 12.6 months, respectively.78 On the basis of these encouraging data, a uveal melanoma-specific phase II trial comparing cabozantinib with temozolomide has been initiated (NCT01835145).
More recently, tremelimumab, a fully human monoclonal antibody that inhibits CTLA-4, offered a median OS of 12.8 months in a phase II trial of 11 patients with advanced uveal melanoma. However, the median PFS of 2.9 months, corresponding to a 6-month PFS rate of 9.1%, and lack of responses led to the trial being stopped early because of futility.80
Adoptive T-cell therapy has shown promise in the treatment of metastatic solid cancers and activity in a uveal melanoma preclinical mouse model.81 ,82 A phase II study (NCT01814046) is investigating the use of chemotherapy followed by autologous tumour-infiltrating lymphocytes with or without high doses of the growth factor interleukin-2 in patients with metastatic ocular melanoma.
Liver-targeted treatment options
The liver is the organ most commonly affected by metastatic disease. At the time of death, approximately 90% of patients have liver metastases; of these, 50% of patients appear to have metastases exclusively in the liver.83
Although evidence is limited, preliminary data suggest improvements in patient outcomes following hepatic resection,84 regional chemotherapy such as hepatic intra-arterial (HIA) chemotherapy85 and hepatic arterial chemoembolisation.86 Retrospective case studies suggest that surgical resection may be curative; however, these studies report on highly selected patient populations and surgery is not suitable for >90% of patients with liver metastases.87 ,88 A single trial has compared the efficacy between HIA and intravenous fotemustine. Although there was no OS benefit (median 14.6 months for HIA vs 13.8 months for intravenous fotemustine), improvements in PFS (4.5 vs 3.5 months) and response rate (10.5% vs 2.4%) were observed with HIA versus intravenous chemotherapy.85
Isolated hepatic perfusion (IHP) involves surgically isolating the blood supply to the liver to allow direct delivery of high-dose chemotherapy. In a trial of 34 patients with isolated liver metastases, the median OS with IHP was 24 months, corresponding to a potential survival benefit of 14 months compared with retrospective controls (p=0.029).89 A phase III trial comparing IHP with the best alternative care in uveal melanoma is underway (NCT01785316). A non-surgical alternative to IHP is percutaneous hepatic perfusion (PHP). A phase III trial of PHP with melphalan compared with best alternative care in 93 patients with ocular (88%) or cutaneous (12%) melanoma demonstrated significantly improved hepatic PFS and overall response rate with PHP therapy. OS was similar between the two arms; however, this was confounded by crossover.90
Localised radioembolisation using yttrium-90 (90Y)-labelled microspheres has also shown benefit for patients with liver metastases. In a small trial of 13 patients, partial responses and stable disease were observed in eight and two patients, respectively.91 The combination of 90Y-labelled microspheres with sorafenib is being studied in a phase I trial (NCT01893099) and combination with ipilimumab is being assessed in a phase 0 study (NCT01730157).
Systemic adjuvant therapy
Adjuvant therapy has not been widely studied in uveal melanoma and there is little evidence that any approach improves patient outcome. However, new approaches to adjuvant therapy are being developed, including rational use of existing cytotoxic and immunotherapeutic regimens using tumour genetic criteria to identify patients at risk. The use of systemic therapy in the adjuvant setting has been discussed in excellent prior publications and the readers are referred to these reviews for additional information.32
Management and surveillance for metastatic uveal melanoma
In most cases of uveal melanoma, patients are diagnosed by an optometrist or ophthalmologist, with subsequent referral to a specialist ocular oncologist. Delays in treatment at this stage can have implications on preventable morbidity and overall patient outcomes. For example, a greater percentage of patients in the UK who experienced delays, or who reported their tumour as being misdiagnosed, ultimately required enucleation.5 Delays may arise from slow referral times, misdiagnosis or failed detection of tumours, delays in imaging and administrative delays.5 ,92
Given the frequent development of metastases, consideration of active surveillance and management of patients following adjuvant therapy has been recommended within the UK Uveal Melanoma National Guidelines.40 Although no survival benefit as a result of the early detection of asymptomatic disease has been documented, surveillance allows for the identification of oligometastatic disease amenable to surgery or other local therapies, as well as patients eligible for clinical trials.39 Risk stratification by clinical criteria, cytogenetic studies and gene expression profiling can be used to identify patients at high risk of developing metastases and guide surveillance decisions accordingly.14 ,37 ,40
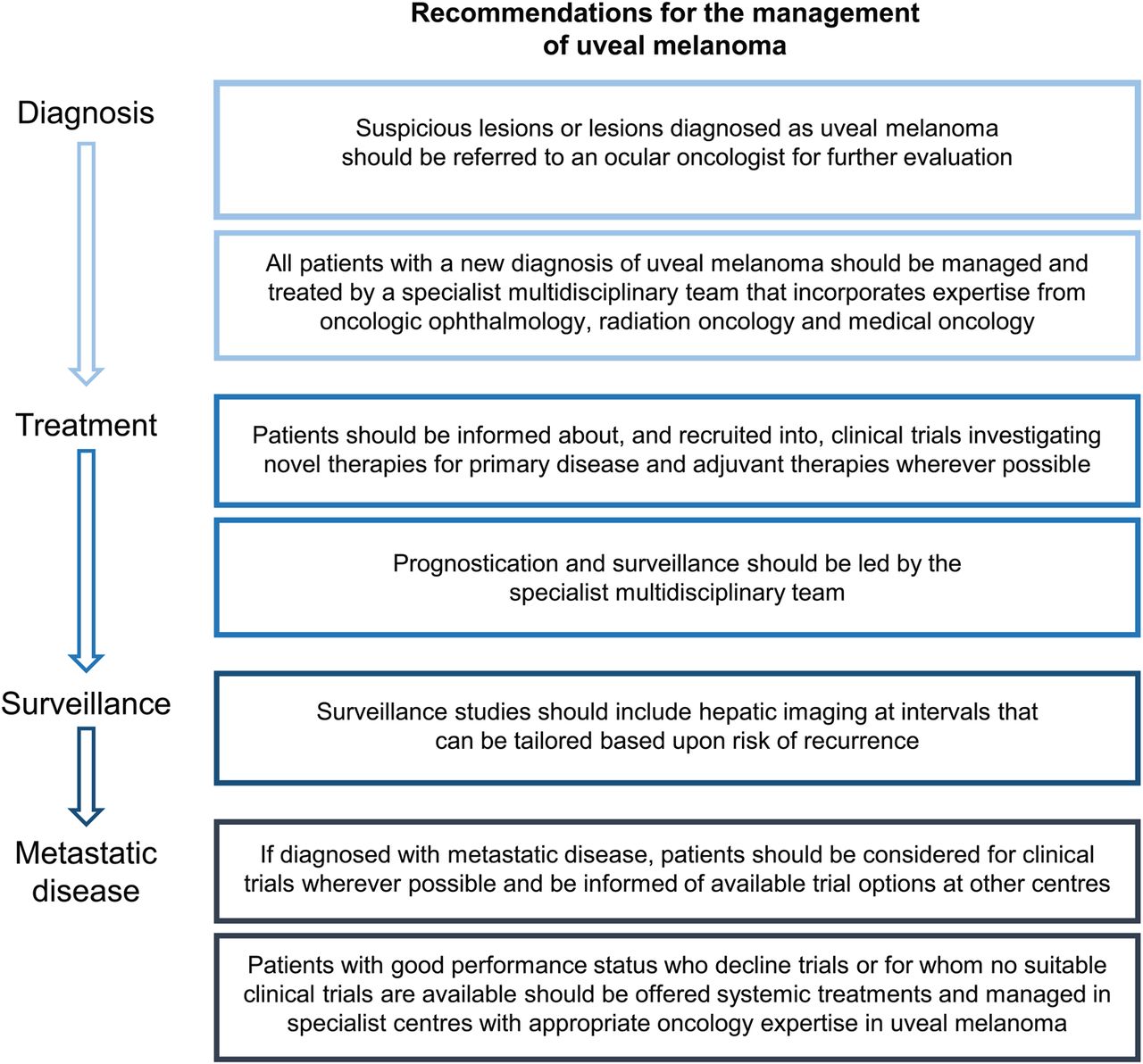
The UK guidelines emphasise the importance of consulting the patient at all stages of their treatment, ensuring individuals are fully informed of any risks, benefits and quality of life implications throughout their treatment and post-treatment surveillance.40 ,93 They recommend that this consultation process should be coordinated by a multidisciplinary team with experience in uveal melanoma treatment, made up of a medical or clinical oncologist, an interventional radiologist, a histopathologist, a liver surgeon and a clinical nurse.40 (figure 2).

{kind=link}
{kind=link}
Summary of key points relevant to multidisciplinary team management of uveal melanoma from the UK Uveal Melanoma National Guidelines.40
Summary
Uveal melanoma is considered rare, but is in fact the most common primary intraocular malignancy in adults. Biologically distinct from cutaneous melanoma, there are a number of underlying somatic gene alterations in uveal melanoma associated with a variable prognosis, which are currently being targeted in clinical drug development. Metastatic disease is associated with a poor prognosis and as such the early identification of patients at high risk of metastases by genetic analysis may allow for personalised management and surveillance. As our understanding of the biology and treatment of this disease develops, multidisciplinary team care will be central to patient management to help optimise outcomes.
Acknowledgments
The authors thank Jon Moran, PhD, from iMed Comms, who provided medical writing support funded by AstraZeneca.
References
Footnotes
Contributors Defining the concept: RDC. Drafting and critical revision: RDC, GKS, TT, BM, JHF and PDN.
Funding AstraZeneca.
Competing interests RDC received non-financial support from AstraZeneca, during the conduct of this work; RDC has received grants, personal fees and non-financial support from AstraZeneca, personal fees from Janssen, personal fees from Thompson Reuters, personal fees from Merck, personal fees from Biogen Idec, personal fees from Aura Biosciences, grants from Melanoma Research Foundation, grants from Melanoma Research Alliance, grants from NIH and grants from ASCO, outside the submitted work. PDN reports personal fees from AstraZeneca, outside the submitted work.
Provenance and peer review Not commissioned; externally peer reviewed.