Article Text

Abstract
Background Methylmalonic acidemia (MMA) and propionic acidemia (PA) are rare hereditary disorders of protein metabolism, manifesting early in life with ketoacidosis and encephalopathy and often resulting in chronic complications. Optic neuropathy (ON) has been increasingly recognised in both conditions, mostly through isolated case reports or small cases series. We here report the clinical features and visual outcomes of a case series of paediatric patients with a diagnosis of MMA or PA.
Methods Retrospective observational case series. A database of patients attending the Willink Biochemical Genetics unit in Manchester was interrogated. Fifty-three patients had a diagnosis of either isolated MMA or PA, of which 12 had been referred for ophthalmic review.
Results Seven patients had clinical findings compatible with ON. Visual outcomes in these patients were poor, with slow clinical progression or stability over time in five cases with follow-up. Presentation was acute in a context of metabolic crisis in two of the cases. Four patients with ON had electrodiagnostics showing absent pattern evoked potentials, with one showing a preserved flash response. All four showed marked attenuation of the dark-adapted electroretinogram with better preservation of the light-adapted response.
Conclusions Our study suggests that ON is under-reported in patients with MMA and PA. Clinical presentation can be acute or insidious, and episodes of acute metabolic decompensation appear to trigger visual loss. Photoreceptor involvement may coexist. Active clinical surveillance of affected patients is important as comorbidities and cognitive impairment may delay diagnosis.
- Electrophysiology
- Optic Nerve
- Child health (paediatrics)
- Genetics
- Vision
Statistics from Altmetric.com
Introduction
Methylmalonic acidemia (MMA) and propionic acidemia (PA) are two of the most common organic acidemias (OA). They are inherited defects of the catabolism of propionate, a common intermediate product of the catabolism of branched-chain amino acids and odd chain fatty acids, caused by variable deficient activity of two mitochondria-located enzymes: methylmalonyl-CoA mutase in MMA and propionyl-CoA carboxylase in PA. These two enzymes consecutively intervene in the conversion of propionate into succinate, which is then fed into the Krebs cycle to produce energy for the mitochondrial respiratory chain. Deficiencies of these enzymes result in the accumulation of intermediate upstream products: methylmalonate and propionate, respectively, alongside other toxic derivatives.
Patients with MMA and PA usually present in the neonatal period with acute metabolic distress (AMD) and encephalopathy, but may present later in infancy in less severe deficiencies with recurrent ketoacidosis, psychomotor retardation and chronic vomiting. Treatment is based on a strict low-protein diet to limit enzymatic substrate, sufficient caloric intake, L-carnitine and antibiotics to reduce intestinal odd-chain, fatty acid-producing bacteria. Stress, infections and inadequate diet can trigger AMD. Despite significant therapeutic improvements over the last two decades, global outcome of patients with OA remains poor, with chronic complications remaining common and progressive.1–3
Although these enzymes are expressed ubiquitously, the clinical features observed indicate a tissue-specific vulnerability (brain, muscle, pancreas, kidney). Both conditions, but especially PA,1 often manifest with encephalopathy, causing permanent damage in the form of variable developmental delay and movement disorders, with frequent basal ganglia lesions on MRI and acute ‘stroke-like’ deficits. MMA also results in renal impairment in the first or second decade of life, whereas cardiac anomalies are common in PA.
Optic neuropathy (ON) is an increasingly recognised complication in the course of both acidemias, but few case reports or series of cases in the literature describe it.4–7 We aimed to define the clinical features and electrodiagnostic findings of paediatric patients with MMA or PA.
Materials and methods
This study comprises a retrospective observational case series and literature review. The database of patients followed in the Willink Biochemical Genetics Unit with a diagnosis of PA or MMA was interrogated, yielding a total of 53 patients (35 with MMA and 18 with PA). Patients who had undergone ophthalmic examination were included for analysis. A literature search (keywords: methylmalonic acidemia, propionic acidemia, organic acidemia, optic neuropathy, optic atrophy, eye) identified previously published reports and case series of PA or MMA with ON.
Results
A total of 12 patients had available ophthalmic records. Of these, seven children had fundus changes (optic atrophy or pallor) and reduced visual acuities compatible with ON, whereas the remaining five did not show evidence of ON (normal optic nerve appearance and/or normal visual acuity (VA)). Clinical observations are summarised in table 1 for patients without visible ON and in table 2 for patients with ON.
Clinical features of patients without signs of optic neuropathy
Clinical features of patients with signs of optic neuropathy
In the group of patients without ON (table 1), one patient (patient 2) had esotropia, ocular apraxia and nystagmus but no fundal evidence of ON. Their pattern visual evoked potential (VEP) was extinguished, but the flash response was normal. Another patient (patient 1) showed delayed pattern VEP, but normal fundoscopy and visual function. In the remaining three, electrodiagnostics were not performed: of these, one child had esotropia but normal fundoscopy (patient 5) and two did not have abnormal ocular findings.
Ages in children with visible ON (table 2) ranged between 6 and 14 years, with a mean of 10 years. There was a male preponderance, with five males and two females; of three patients with PA, two were male and one female. Out of four patients with MMA, three were male and one female. All showed a degree of optic nerve pallor that was subtle in two cases (8 and 10) and severe in the remaining five, with unremarkable retinal examination.
Clinical presentation of ON was progressive, insidious or undetermined in five children, and acute or subacute in two. Three had strabismus on examination, and one reported temporary esotropia during previous illness. Of those who presented insidiously, ON was detected in routine eye referrals for esotropia in two. Two children were referred with a history of long-standing visual difficulties and in another child the onset of visual loss could not be determined (figure 1).

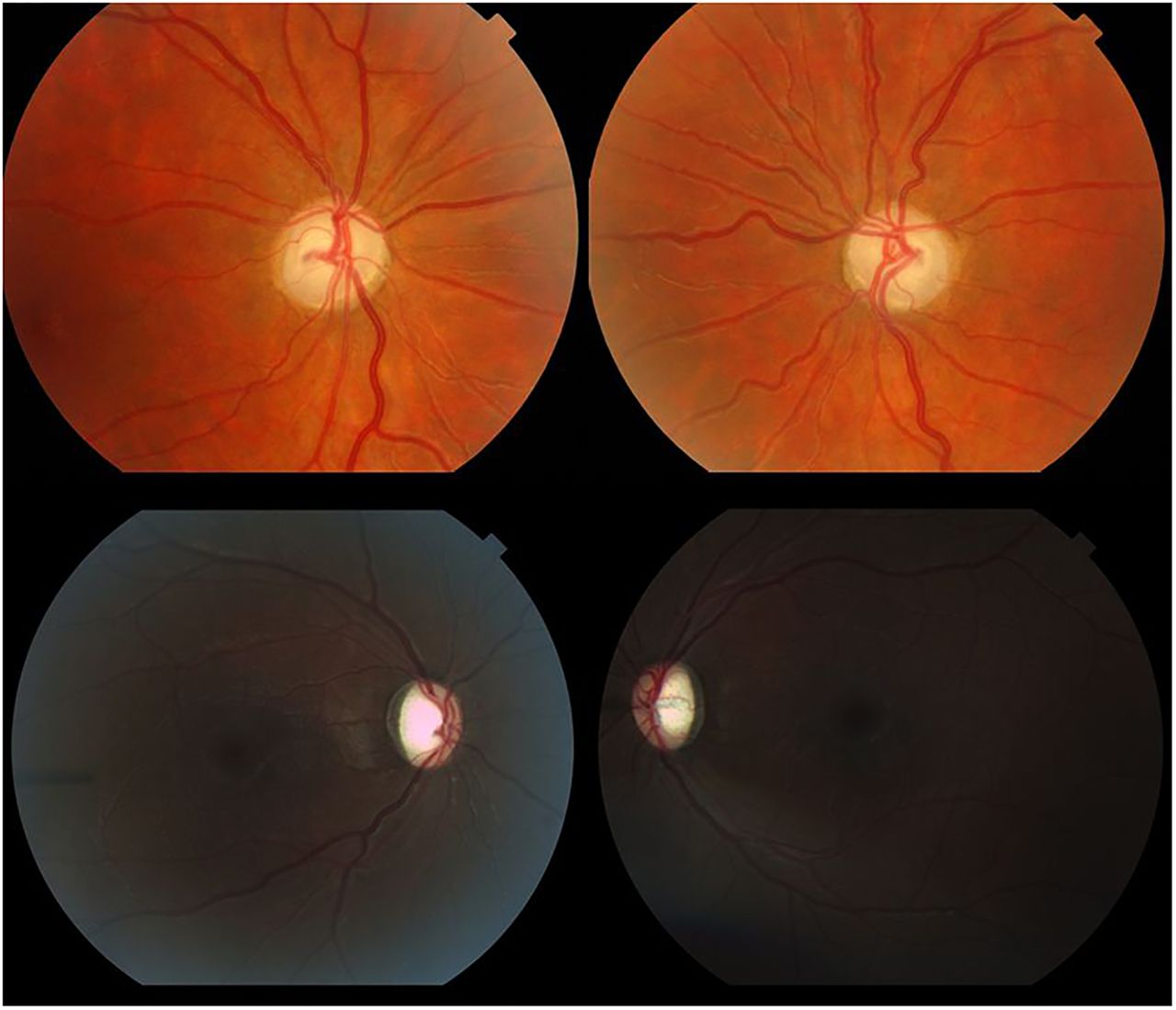
Top: Fundal pictures show diffuse optic nerve pallor in a 14-year-old male (case no. 6 in the table) with methylmalonic acidemia of neonatal onset, complicated with end-stage renal failure necessitating transplant. Final visual acuity was light perception in the right eye and 6/48 in the left. Bottom: Bilateral optic atrophy in an 11-year-old female (case no. 12 in the table) with propionic acidemia diagnosed days after birth. Marked optic cupping is observed, found to have developed 24 months after acute bilateral loss of vision during a metabolic crisis. Initial cup/disc ratio was 0.2 for either eye. Visual acuity was hand movements and 6/60 for right and left eyes, respectively. Both cases had electrodiagnostics, showing absent evoked potentials and decreased dark-adapted responses on electroretinography. RNFL, retinal nerve fibre layer.
Two children (cases 8 and 12) presented acutely with complaints of visual loss during episodes of AMD that required long hospital admissions and intensive care with other concomitant complications such as pancreatitis (case 12), and worsening spasticity in case 8, both requiring ventilation and tracheotomy (figure 2). Case 8 only had partial visual improvement following stabilisation. A third patient (case 6) had acute worsening of his VA during AMD secondary to an episode of posterior reversible encephalopathy in a context of renal failure and hypertension.
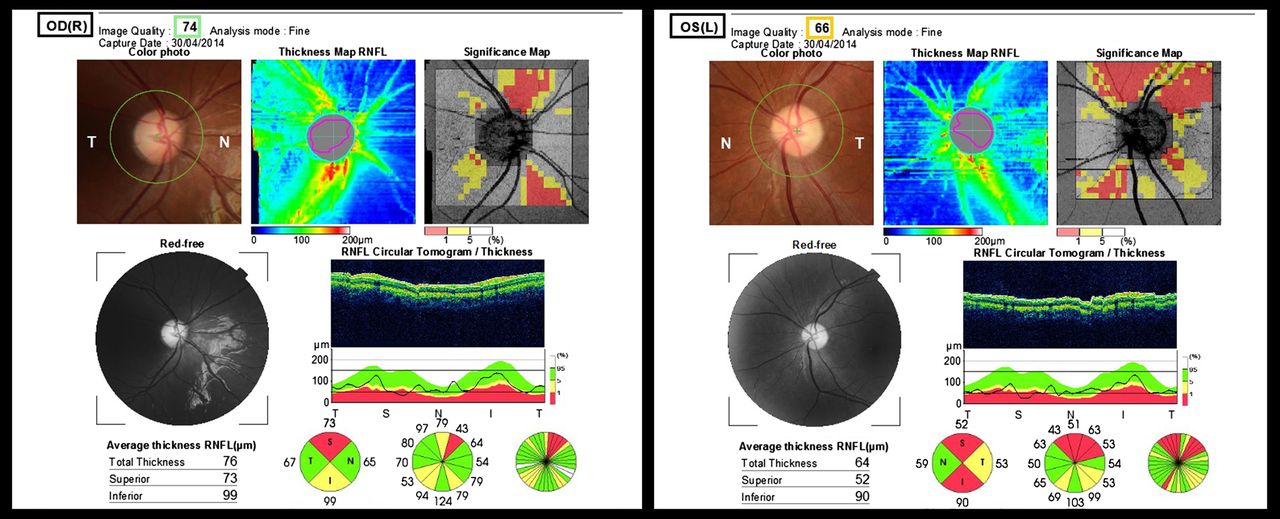
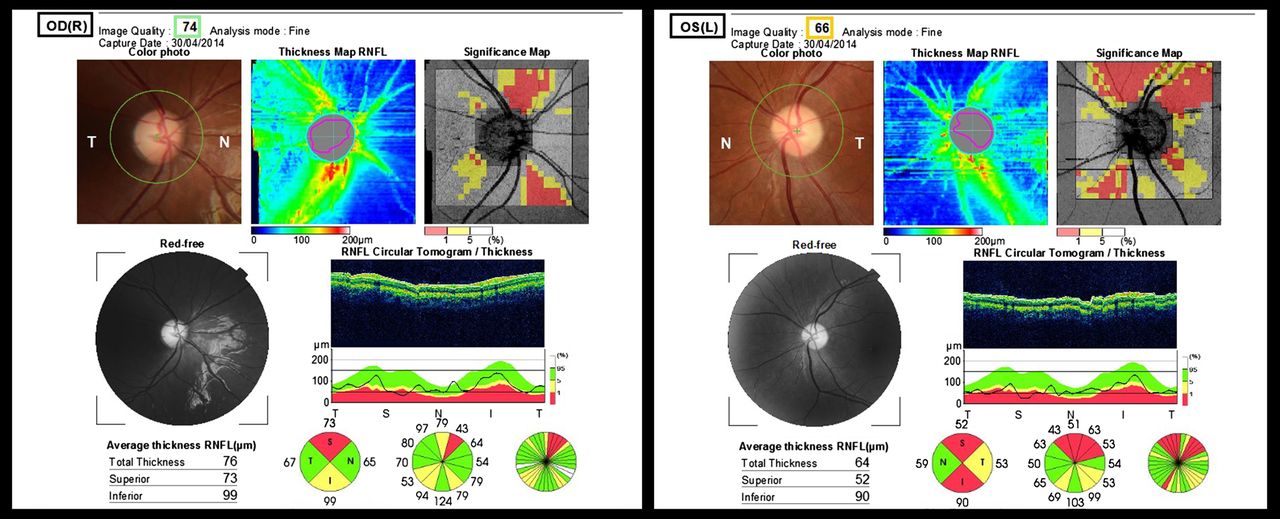
Topcon optical coherence tomography (OCT) of the retinal nerve fibre layer and fundal pictures of an 11-year-old female (case no. 8 in the table) with methylmalonic acidemia. Diagnosed as a neonate, previous complications included severe renal failure and extrapyramidal movement disorders following earlier basal ganglia infarction. She presented with subacute bilateral decrease of visual acuity recorded at 6/60 for right and left eyes and bilateral dyschromatopsia, noted concomitant to a severe episode of metabolic decompensation with initial normal fundoscopy and mildly subnormal acuities. Four months later, fundal pictures show bilateral disc pallor with asymmetric retinal nerve fibre layer thinning of 76 μm in the right eye and 64 μm in the left on OCT. At the time of the OCT, visual acuity had improved and was 6/9.5 and 6/30 for right and left eyes, respectively.
VA at presentation of ON was worse than 6/60 (Snellen) in five eyes, between 6/60 and 6/30 in seven eyes and only two eyes were better than 6/30 (table 2). Follow-up after diagnosis was available in five children, ranging between 12 and 48 months, with a median of 20 months. All deteriorated or were stable over time, except case 8, who experienced a partial improvement in VA. Final VA was worse than 6/60 for four eyes, between 6/48 and 6/30 in four eyes and better than 6/30 in two.
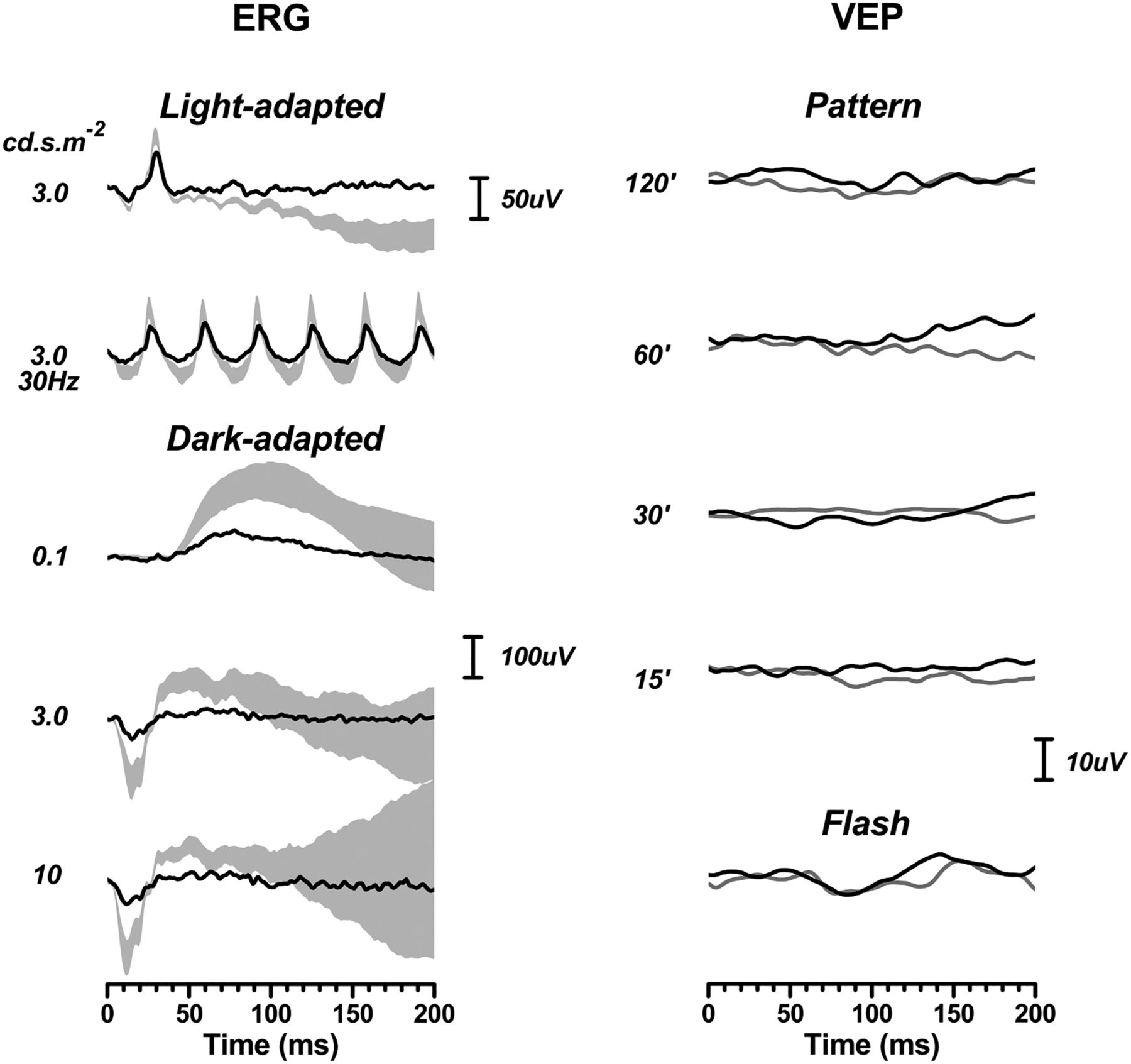
Four children had International Society for Clinical Electrophysiology of Vision standard electrodiagnostics (cases 6, 7, 10 and 12). Pattern VEPs were absent in all four, as were flash responses in three of them. All four showed marked electroretinogram (ERG) attenuation, particularly in the dark-adapted state (by about 80%), but also to some extent (about 50%) in the light-adapted state (figure 3).

{kind=link}
{kind=link}
{kind=link}
Electrodiagnostics, from case 12, showing absent flash and pattern flash visual evoked potentials (VEPs) (right column). Electroretinogram (ERGs) were recorded in cases 6, 7 and 12 using corneal fibre electrodes and complied with international standards. Case 10 had ERGs recorded using skin electrodes, for reasons of compliance. Only case 12 is shown because the findings from the three other patients are remarkably similar. The light-adapted (cone) responses are attenuated to some extent but this is much greater in the dark-adapted state, suggesting a rod-mediated retinal dysfunction. The shaded areas in the ERG depict 95% CIs from our normative data set. The VEP data show 2 responses per condition.
Out of the four children with MMA and ON, all had some degree of renal failure and neurological involvement at diagnosis. Renal failure was severe in three, necessitating a renal transplant in two. Two had MRI evidence of damage to the basal ganglia and another had leg spasticity; one had behavioural problems and other, mild learning difficulties. In the three children with PA and ON, two had significantly prolonged Q-T intervals. Two had developmental delay, along with epilepsy in one case, and other showed MRI changes in the basal ganglia; the third child had generalised myopathy. B12 levels were measured in several occasions during follow-up under the metabolic unit, and these were within normal limits.
Discussion
From our database of 53 patients with MMA and PA, 12 patients underwent ophthalmic review and 7 of these had clinical signs of ON, suggesting an incidence of ON of at least 13% in these patients. Two of the five patients without clinical signs of ON also had subnormal VEPs indicating subclinical involvement.
Other authors have reported a visual impairment rate of 7% in OA, with no details provided regarding the aetiology of the visual loss.2 Our findings suggest that the incidence of visual impairment is likely to be higher. Whereas five patients with ON were referred with a complaint of visual difficulties or visual loss, two were found to have clinical ON and a further two subclinical ON during ophthalmic screening, and this seems to also have been the case in at least two other paediatric patients reported in the literature.4 The remaining 41 patients under metabolic follow-up that were not referred to our department for examination were not recognised as having visual impairment. However, the very common coexistence of developmental delay and other severe chronic, life-threatening complications would be potential factors contributing to under-diagnosis.
Previously reported ocular findings in PA and MMA have included ON, cataracts and ocular apraxia. A literature search identified 11 patients with MMA or PA with a diagnosis of ON.4–9 Details of these patients are summarised in table 2. A distinct subtype of MMA associated with a specific deficiency in the cobalamin metabolism (CblC subtype) is known to manifest with a prominent maculopathy and retinopathy, which has been well described in the literature.10 We have excluded these patients from our review and database search due to their different ocular and systemic phenotype.
The mean age of patients previously reported with MMA and PA and optic atrophy was higher than our series (15.5 years), with ranges between 2 and 24 years old. Those who were older at diagnosis were better characterised clinically, with acute bilateral loss of vision in six, and sequential visual loss in two. Perimetry showed centro-cecal scotomas in four reported cases and a concentric scotoma in one patient. Those who were younger (13 years old or less) were either detected in screening4 or presentation was undetermined. Visual outcome for 20 eyes was poor with a VA below 6/60; only one patient had a better outcome (patient 4 in table 3), who had fluctuating vision in the previous months and a partial recovery after starting antioxidant treatment. Two patients had VEP, and both showed delayed responses.
Clinical features of reported cases of optic atrophy in methylmalonic and propionic acidemia
In our patients, and in the global group, presentation of the ON was variable, though all were bilateral and relatively symmetric. Older patients in the literature have mostly presented acutely with centro-cecal defects, temporal pallor and ultimately profound bilateral visual loss that has been compared with Leber's hereditary optic neuropathy (LHON).5 ,7 ,8 Younger patients in the literature, and the five males in our paediatric series, presented with progressive or undetermined visual deterioration, which may in part reflect the younger age and increased difficulty in reporting visual changes.
These similarities with mitochondrial ON are supported by the suspected pathophysiology of some of the complications. Mitochondrial dysfunction (primary or secondary to mitochondrial metabolite accumulation) is believed to play an important role, particularly underlying acute neurological symptoms (stroke-like episodes).2 ,3 The observation of clinical features shared with mitochondrial disease (MD) and the biochemistry abnormalities during metabolic crisis further support this association. Currently, a number of in vitro, postmortem and murine model studies show strong evidence of mitochondrial malfunction in these patients.11–16 Murine models have suggested that both mitochondrial dysfunction and direct metabolite toxicity are synergistic in causing neural tissue damage in MMA.17
Other ON such as toxic-nutritional and toxic (eg, ethambutol-induced) can also result in selective initial damage to the papillomacular bundle with some scope for reversibility, especially if the optic atrophy is not established at diagnosis. This can also be true in LHON, which can show a degree of spontaneous recovery in some mutations.18 While the cases of acute presentation would be reminiscent of LHON, those that are insidious would be clinically more similar to other MD such as dominant optic atrophy (DOA), toxic or nutritional ON. Patient 10 in the literature series developed sensorineural hearing loss 3 months after presentation, and patient 3 had bilateral hearing loss. De Baulny et al2 mentioned two further patients with optic atrophy and sensorineural hearing loss, with no other details provided. In patients with other forms of MD or toxic neuropathy, both sensorineural hearing loss and ON are sometimes associated (eg, 20% of families with DOA develop hearing loss, usually occurring later in life).19
Nutritional factors may also contribute to the ON in patients with MMA and PA. Poor weight gain due to protein restriction and anorexia are very common, often requiring gastrostomy tubes to supply part or all of the daily calories. This could contribute to the ON in a similar way as in toxic-nutritional ON, with poor nutrition overlying a context of oxidative stress and toxic metabolites. Three of the patients reported in the literature had B group vitamins measured after vision loss, with only one showing slightly low levels of B1 and B6.7 ,8
Antibiotics are used in these patients to reduce the amount of propionate produced by gut bacteria, metronidazole being part of the treatment protocol for MMA and PA;1 all of our patients received lifelong treatment with this drug. While reversible ON has been reported in one patient after treatment for 8 months on metronidazole,20 peripheral autonomic, motor and sensory neuropathies are recognised side effects, particularly in high doses and long-term treatments.
Acute neurological and other complications in MMA and PA happen often during or shortly after recovery from a metabolic crisis.2 In our group, two of the children presented while suffering AMD, with three patients reported in the literature mentioning a recent AMD prior to vision loss.7 ,9 ,11 In patient 8 of our series with MMA and AMD, VA improved spontaneously (without specific antioxidant treatment) after metabolic recovery and patient 12 received coenzyme Q10 (CoQ10) while in hospital, with no recovery in vision.
Three of the literature cases also had treatment trials with antioxidants: CoQ10 in one case and a combination of CoQ10 and vitamin E in the other two. One case treated with CoQ10 and vitamin E, started 2 months after the vision loss, had visual improvement.6 In another patient, only CoQ10 was administered, and in other the combination was given 7 months after onset, both with no benefit.7 ,8
Isolated administration of CoQ10 is being investigated as a treatment option for LHON; class I evidence has shown that it can improve outcomes in certain subtypes.19 Recent investigation on a murine MMA model showed that an oral combination of CoQ10 and vitamin E improved the rate of decline of glomerular filtration rate in two comparable groups of MMA mutant mice fed with high protein concentration.21 This experimental evidence, alongside the anecdotal evidence, would support the opening of research into future treatment options for ON in PA and MMA.
Ianchulev et al4 suggested that optic atrophy in PA may have a preponderance for males. Out of all the reported cases for both conditions (our series included), there was a male preponderance of 2:1 with 12 males and 6 females; 5 males/3 females for MMA and 7 males/3 females for patients with PA. Given the small amount of published cases, it is difficult to know if this has any significance. Of the only two patients in this series with visual improvement, both were females and had MMA; one had early treatment with CoQ10 and E vitamin,6 while the other (patient 8) improved with only metabolic stabilisation.
All four patients with clinical ON who had electrodiagnostics had very reduced or absent VEP as would be expected in advanced optic atrophy, but interestingly also had grossly reduced dark-adapted ERGs, with relative preservation of the light-adapted response. They all had established visual loss and poor acuities—worse than would be expected from the ERG alone—pale nerves and unremarkable retinas on fundoscopy. Photoreceptor dysfunction has not been previously reported in MMA (excluding CblC subtype) or PA, with only delayed VEP reported6 ,8 and should be investigated further. A similar ERG could be found in coexistent vitamin A deficiency, which was not tested for, though a toxic cause or a mitochondrial dysfunction-mediated occult retinopathy, such as are presumed to cause ON and other complications, would be potential factors. Visual fields were not performed in our patients.
In conclusion, our series suggests that the incidence of ON with severe visual impairment is significant in MMA and PA. Clinical presentation is variable, with progressive and sudden onset being possible. Periodic ophthalmological screening is therefore important to determine the presence of visual impairment, particularly in children and in patients with developmental delay. As the treatment modalities and survival of these patients improve, the recognition of this complication is likely to increase.
More dedicated studies are needed to identify the prevalence of ON in these conditions to determine the extent and prevalence of photoreceptor involvement and determine the possible influence of nutritional deficiencies. Based on clinical observation, experimental and postmortem evidence, the aetiology of ON in these patients seems likely to stem from mitochondrial malfunction; however, a multifactorial aetiology remains possible.
Acknowledgments
This study was facilitated by the Manchester Biomedical Research Centre and the Greater Manchester Comprehensive Local Research Network. We are grateful to Claire Delaney, Lis Nichol and Lindsi Williams for the electrodiagnostic workups. Wai Chan contributed to the original concept for this manuscript.
References
Footnotes
Contributors All co-authors listed in this article have been directly involved with either data collection, review of literature or preparation of the manuscript.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.