Article Text

Abstract
Background To evaluate the safety and tolerability of ranibizumab 0.5 mg in patients with uni/bilateral neovascular age-related macular degeneration (nAMD) and best-corrected visual acuity (BCVA)<2/10 and/or second eye affected, regardless of BCVA.
Methods In this 12-month, prospective, multicentre, open-label, single arm, pragmatic interventional study, patients (N=941) aged ≥ 50 years were to receive ranibizumab as per approved label, monthly until maximum stable visual acuity (VA) was achieved (initially, three or more injections may be required). Thereafter, patients were to be monitored monthly for VA and treatment was to be resumed if VA was reduced due to disease activity.
Results Of the 936 patients treated with ranibizumab at least once during the study, 823/113 were unilaterally/bilaterally (not simultaneously) treated . The mean (SD) number of ranibizumab injections during the study was 5.4 (2.9)/10.6 (5.0) injections in uni/bilaterally treated patients. Three systemic drug-related adverse events (AEs) (all serious, all in unilaterally treated patients) and 18 systemic AE of special interest (AESIs) (11 serious, 16/2 in unilaterally/bilaterally treated patients) occurred during the study. The annual incidence rate (AIR) (events/1000 person-years) for systemic drug-related AEs, considering a 15-day/30-day risk period, 11.0/8.5 for unilaterally treated patients. Considering the same risk period, the AIR (events/1000 person-years) for systemic AESIs for unilaterally treated patients was 22.1/19.9. Considering a 30-day risk period, the AIR (events/1000 treated eye-years) of ocular drug-related AEs was 23 and AESIs was 11.5.
Conclusions The low incidence of AEs and AESIs demonstrated the good safety and tolerability of ranibizumab in unilaterally/bilaterally treated patients with nAMD in this real-world setting.
- neovascularisation
- retina
- vision
- age-related macular degeneration
- unilateral AMD
- bilateral AMD
- neovascular age-related macular degeneration
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- neovascularisation
- retina
- vision
- age-related macular degeneration
- unilateral AMD
- bilateral AMD
- neovascular age-related macular degeneration
Introduction
Age-related macular degeneration (AMD) affects nearly 8.7% of the worldwide population, and the numbers are projected to increase to around 196 million in 2020.1–6 In Europe, projections based on a recent meta-analysis of AMD prevalence data using the European Eye Epidemiology (E3) consortium show an almost doubling of patients with AMD by 2040, to between 14.9 and 21.5 million with early AMD, and between 3.9 and 4.8 million with late AMD.7 In Italy, the PAMDI study showed that AMD affects a high percentage of the elderly population; of the 1162 patients aged ≥61 years included in the study, 62.7% had AMD.8
AMD is one of the most common causes of permanent central vision loss in the older population aged ≥65 years, predominantly in developed countries.4 9–12 Neovascular AMD (nAMD) accounts for majority of the AMD-associated vision loss, even though it accounts for only 10%–20% of the overall cases of AMD.12 13
Antivascular endothelial growth factor (VEGF) agents are the current standard of care for the treatment of visual impairment due to choroidal neovascularisation (CNV) secondary to nAMD.12 Ranibizumab 0.5 mg (Lucentis®; Novartis Pharma AG, Basel, Switzerland, and Genentech, South San Francisco, California, USA) was the first anti-VEGF agent to be approved for this indication, in many countries, based on the results from two pivotal trials: Antibody for the Treatment of Predominantly Classic Choroidal Neovascularization in Age-Related Macular Degeneration (ANCHOR)14 and Minimally Classic/Occult Trial of the Anti-VEGF Antibody Ranibizumab in the Treatment of Neovascular Age-Related Macular Degeneration (MARINA)15 studies. The burden of the second eye developing nAMD is so high that nearly 50% of all eyes at risk would require bilateral treatment by 3 years.16 However, there are limited data on treatment outcomes in the second-affected eyes, considering most clinical trials include one study eye per patient, to minimise bias and for statistical reasons.17
In Italy, after the authorisation of ranibizumab for nAMD in 2007,18 limitations were applied on reimbursement. Patients with visual acuity (VA) <2/10 were excluded and for those patients with bilateral disease, the treatment of one eye only was reimbursed;19 the physician decided which eye to treat, usually the eye with better vision. These patients were therefore excluded from the Lucentis monitoring registry at the Italian Medicines Agency (AIFA) that tracks patients’ eligibility and evaluates the appropriateness of treatment in the approved indications, as well as collecting safety information useful to AIFA to integrate the risk-benefit profile of the drug with data from clinical practice. However, following the approval of ranibizumab 0.5 mg in new indications in 2012, the limitations on the reimbursement for patients with VA <2/10 and those with bilateral disease were lifted; hence, the AIFA wanted to assess the safety of ranibizumab 0.5 mg in these specific subgroups.
The TWEYEs study (group members of this study are listed in online supplementary appendix 1) was specifically conducted to address this gap, and evaluated the safety and tolerability of ranibizumab 0.5 mg in patients with a VA of <2/10 and those with bilateral nAMD, in a real-life setting in Italy.
Supplemental material
Materials and methods
Study design
TWEYEs was a 12-month, prospective, multicentre, open-label, single-arm, pragmatic interventional clinical study that was conducted across 107 centres in Italy, from March 2014 to June 2016.
The study protocol was reviewed and approved by the Ethics Committee (EC) of each site and the study was conducted in accordance with the Declaration of Helsinki and applicable local regulatory requirements. Patients provided written informed consent at screening. The study is registered with Clinicaltrials.gov (identifier, NCT01986907).
Patients
Patients aged ≥50 years were included if they fulfilled the following criteria: eyes with best-corrected VA (BCVA) <2/10 due to nAMD (20/100 Snellen equivalent), in both unilaterally and bilaterally affected patients; fellow eye regardless of BCVA in patients with bilateral nAMD, if the first eye had been treated with ranibizumab at least 14 days before (fellow eyes developing nAMD [newly diagnosed] while the study was running were also included in this category).
Key exclusion criteria were active intraocular inflammation (grade trace or above) in either eye; current or suspected ocular or periocular active infection (eg, conjunctivitis, keratitis, scleritis, uveitis, endophthalmitis) in either eye; evidence of retinal pigment epithelial tear at enrolment or vitrectomy in the study eye; ocular disorders that could have confounded the interpretation of results, compromised VA or required medical or surgical intervention during the study period such as mature cataract, retinal vascular occlusion, pre-retinal membrane (macular pucker), retinal detachment or macular hole; uncontrolled glaucoma (intraocular pressure [IOP]≥30 mm Hg on medication or according to investigator’s judgement) in either eye; systemic treatment with any VEGF inhibitor, including bevacizumab and ziv-aflibercept in the 90 days prior to study enrolment; intraocular treatment with any anti-VEGF or verteporfin photodynamic therapy within 1 month prior to study enrolment; intraocular surgery within 28 days prior to study enrolment; stroke (brain ischaemia), transient ischaemic attack (TIA) or myocardial ischaemia in the 3 months prior to study enrolment; known hypersensitivity to ranibizumab or any other component in the formulation. Pregnant or nursing women and women of childbearing potential not using effective methods of contraception were also excluded.
Study treatment
All patients were to be treated as per the approved ranibizumab label that was in effect at the time of the study (Summary of Product Characteristics [SmPC] 2013)20: Treatment was initiated with one injection per month until maximum VA was achieved and/or there were no signs of disease activity that is, no change in VA and in other signs and symptoms of the disease under continued treatment. In patients with nAMD, initially, three or more consecutive, monthly injections may be needed. Thereafter, patients were to be monitored monthly for VA and treatment was to be resumed in case of VA loss due to disease activity. If the patient was already under treatment with ranibizumab at study initiation, treatment during the study was to be given as per approved label (monthly until maximum VA was achieved).
A patient was considered to be unilaterally treated for the whole period if he/she had only one eye treated. When the second eye was also treated, the patient was considered to be bilaterally treated; however, there was an interval of 14 days between treatment of the two eyes.
Objectives
The primary objective was to evaluate the annual incidence rates (AIR) of ocular and systemic drug-related adverse events (AEs) and AEs of special interest (AESI). The AESIs were those events possibly related to ranibizumab as per the Periodic Safety Update Report (PSUR) No. 13 and Risk Management Plan (RMP) for ranibizumab (online supplementary appendix 2).
Supplemental material
The key secondary objectives included estimating the proportion of systemic and ocular AEs stratifying by type of patient (ie, treated unilaterally or bilaterally) and describing the overall treatment exposure to ranibizumab.
Assessments
Safety assessments included collecting information on any AEs throughout the study.
BCVA was to be assessed at baseline and every month, before any other procedure or drug administration, using Snellen VA testing charts.
Statistical analysis
It was estimated, based on literature data, that in the worst case the observed incidence rates of eye with ocular-suspected treatment-related AEs in this study should be around 1%, as well as the incidence rate of patients with systemic suspected treatment-related AEs.14 21 22 Considering a 30% dropout of patients/eyes and assuming an average of 50% of the observation period (ie, 6 months), with a cohort of 5000 eyes, a total of 4250 eyes/year yielded a two-sided 95% CI of 0.7% to 1.3% for an incidence rate of 1%.
The conservative risk periods related to the ranibizumab injections were defined as 30 days for ocular AEs and both 15 and 30 days for systemic events, based on the vitreous elimination half-life of ranibizumab of about 9 days, intrinsic systemic elimination half-life of approximately 2 hours and the systemic to vitreous exposure ratio of 1:90,000.23 This in turn would have led to reduction of any bias in the study. Based on these premises, conservative risk periods related to the ranibizumab injections were defined as 30 days for ocular AEs and both 15 and 30 days for systemic events. Events occurring outside of these risk periods were considered as occurring in the ‘control period’.
The systemic AIR of drug-related AEs, systemic AIR of AESI/drug-related AEs, ocular AIR of drug-related AEs, and ocular AIR of AESI/drug-related AEs are explained in detail in online supplementary appendix 3.
Supplemental material
The time at risk for systemic events was defined for a 15-day and 30-day period, and the time-at-risk for ocular events was defined for a 30-day period; these have been explained in detail in online supplementary appendix 3.
The safety population consisted of all enrolled patients who signed the informed consent, received at least one dose of study drug and had at least one postbaseline safety assessment. All the AEs occurring from the first injection were considered for the analysis in the safety set independently from the loss of follow-up.
Two subpopulations, systemic safety and ocular safety subpopulations, were introduced in order to evaluate patient safety by means of a sensitivity analysis of the primary objective; details of which are included in online supplementary appendix 3 and online supplementary table 1.
Supplemental material
Patients/eyes were included in each analysis based on available assessments and missing data were not replaced. All statistical analyses were produced using SAS for Windows release 9.4 (64-bit) or later (SAS Institute, Cary, North Carolina, USA).
Results
Patient disposition and baseline characteristics
Of the 944 screened patients, 941 (99.7%) were enrolled and 774 (82.3%) patients completed the study. The main reasons for discontinuation were withdrawal of consent (8.3%) followed by loss to follow-up (4.0%; figure 1). Among the patients that withdrew the consent, there were three patients with serious AE.

Patient disposition. *Patients who were treated with ranibizumab 0.5 mg at least once and five patients did not received ranibizumab treatment. †Percentages are based on the number of patients enrolled (n=941). VA, visual acuity.
Of the 941 enrolled patients, 936 (99.5%) patients were treated with ranibizumab at least once and the remaining five patients did not receive any treatment. All 936 patients were included in the safety set. The ocular safety and systemic safety subpopulations consisted of 908 (97%) and 886 (94.7%) patients, respectively.
At baseline, of the 936 patients included in the safety set, 534 (57.0%) patients had unilateral nAMD, 399 (42.6%) patients had bilateral nAMD and 3 (0.3%) patients were unaffected by nAMD (but had CNV secondary to pathological myopia). During the study, 823 patients (87.9%, all 534 with unilateral nAMD at baseline, 286 with bilateral nAMD and the 3 patients with CNV) were treated unilaterally, 57 (6.1%) were treated bilaterally (both eyes enrolled at the same time), and the remaining 56 (6.0%) were first treated unilaterally and subsequently bilaterally (when the other eye developed the disease).
In the total population, the mean age (SD) of patients was 78.7 (7.3) years; 51.6% of the patients were in the age range of 75–84 years. The majority of the enrolled patients were female (62.2%) and Caucasian (99.9%; table 1). A VA <2/10 was observed in the treated eye in 89.3% of the 823 unilaterally treated (but not necessarily unilaterally affected) patients; and in both treated eyes in 27.4% of the 113 bilaterally treated patients (online supplementary table 2). Seven hundred and seventy-one of the 1049 treated eyes (73.5%) were naïve to ranibizumab at baseline; of these, 600 were unilaterally treated and 171 were bilaterally treated during the study.
Supplemental material
Baseline patient demographics, ocular and disease characteristics (Safety set*)
At least one prior or concomitant medical condition was reported in 902 (96.4%) patients; 792 (96.2%) patients in the unilaterally treated group and 110 (97.4%) patients in the bilaterally treated group (online supplementary table 3). The prior medical condition with the highest incidence was cataract operation (n=235, 25.1%), and the active medical condition with the highest incidence was hypertension (n=579, 61.9%; online supplementary table 3).
Supplemental material
There were few patients who entered the study without satisfying inclusion criteria while many were included who met the exclusion criteria but did not meet the protocol procedure, and label schedule (online supplementary table 4). During the study, over half of the patients (51.9%) were not monitored monthly for VA, and 5 (0.5%) did not receive at least one dose of study drug.
Supplemental material
Treatment exposure
The mean (SD) number of injections per patient in the overall safety population was 6.0 (3.6) and was 5.4 (2.9) in the unilaterally treated patients and 10.6 (5.0) in the bilaterally treated patients. Over the 1-year period, 30.7% of patients received fewer than or equal to three ranibizumab injections, and there was a sharp and continuous decrease in the proportion of patients receiving subsequent injections—4 injections: 11%, 5 injections: 11.5%, 6 injections: 12.6%, 7 injections: 7.9% (online supplementary figure 1). No differences were observed in the mean (SD) number of injections per treated eye in the two groups; 5.4 (2.9) in the unilateral treatment group and 5.3 (2.74) in the bilateral treatment group (overall safety set: 5.3 [2.9]).
Supplemental material
In the treated eyes naïve to ranibizumab at baseline, the mean (SD) number of injections was 4.9 (2.5); 4.9 (2.5) and 5.0 (2.6) in the unilaterally and bilaterally treated patients, respectively.
Annual incidence rate of systemic drug-related AEs and systemic AEs of special interest
A systemic AE was classified under the unilateral or bilateral group according to the current treatment of the patient. Hence, events that occurred in the 56 bilaterally treated patients who were initially treated unilaterally were classified under the unilateral group if they occurred prior to the start of bilateral treatment and under the bilateral group if they occurred after the start of bilateral treatment. For this reason, the sum of unilateral and bilateral patients shown for the primary outcome is greater than the total number of patients.
There were three systemic drug-related AEs reported during the study, all serious, and all occurred in unilaterally treated patients (table 2). The events of angina pectoris and cerebral haemorrhage resulted in study discontinuation. The patient with TIA was lost to follow-up. Two of the three events (TIA and cerebral haemorrhage), occurred within the 15 day risk period and all three occurred within the 30-day risk period.
Systemic drug-related adverse events (by Preferred Term) and systemic adverse events of special interest (by Safety Risk and Preferred Term; Safety set*)
The AIR considering a 15-day risk period was 0.0089 (8.9 cases per 1000 person-years; 95% CI 0.0 to 0.0213)) for the overall population and 0.0110 (approximately 11 cases per 1000 person-years; 95% CI 0.0 to 0.0264) for the unilaterally treated. With a 30-day risk period, the AIR was 7.2 cases per 1000 person-years (95% CI 0.0 to 0.0154) overall and 8.5 cases per 1000 person-years (95% CI 0.0 to 0.0181) for the unilaterally treated (figure 2). No systemic drug-related AEs occurred in the bilaterally treated patients, so it was not possible to calculate AIR for this subpopulation. In the self-controlled case series, with a 15-day and a 30-day risk period, the AIR was 1.5 cases per 1000 person-years (95% CI 0 to 0.0046) and 0 cases (95% CI 0 to 0), respectively (figure 2). The number of events and AIR for the systemic safety subpopulation in sensitivity analyses were comparable with that of the safety population. Systemic AESIs were reported in 18 (1.9%) patients: 16 in unilaterally treated and 2 in bilaterally treated (table 2). Hypertension (n=5), non-myocardial arterial thromboembolic events (n=5) and non-ocular haemorrhage (n=6) were the main risk categories. Eleven of these events were serious. Seven events (traumatic haematuria, blood pressure increase, blood pressure fluctuation, hypertension, epistaxis, varicose vein rupture) were non-serious (table 2). The five patients who had non-myocardial arterial thromboembolic events (cerebral haemorrhage [n=1], cerebrovascular haemorrhage [n=1], and TIA [n=3]) received 1–5 injections prior to the event and were between 77 and 89 years of age; all had concurrent hypertension and cardiopathy. Of the five patients, four were naïve to ranibizumab treatment at baseline. Two of these events (cerebral haemorrhage and TIA) were suspected by the investigator to be related to treatment.

Annual incidence rates of systemic drug-related AEs and AESIs—15-day and 30-day risk periods* (Safety set†). *Time at risk was defined as the sum of the 15 days following each ranibizumab injection. If a patient had more than one injection, his/her risk period was defined as: (1) Time elapsed from the injection to the 15 subsequent days—for injections given more than 22 days from the previous; (2) Time elapsed from the first injection to the 15 days following the last injection—for injections given up to 22 days from the previous one. The same analysis was performed considering a time window of 30 days. †Consisted of all enrolled patients who signed the informed consent, received at least one dose of study drug and had at least one postbaseline safety assessment. Lower limits less than 0 were set to 0. In the bilaterally treated patients, no systemic drug-related AEs occurred during the study, and no systemic AESIs occurred during the 15-day and 30-day risk periods, so it was not possible to calculate AIR for this subpopulation. AE, adverse event; AESI, AE of special interest.
Four events (haematuria, TIA, epistaxis, and cerebral haemorrhage [all except epistaxis were serious]), all in unilaterally treated, occurred within the 15-day risk period, corresponding to an AIR of approximately 18 events per 1000 person-years (95% CI 0.0004 to 0.0353; figure 2). Seven events (five serious [haematuria, TIA, epistaxis, cerebral haemorrhage, increased blood pressure, pulmonary embolism and intestinal haemorrhage] and two non-serious [epistaxis and increased blood pressure]), all in unilaterally treated, occurred within the 30-day risk period (table 2). The AIR was 16.9 per 1000 person-years (95% CI 0.0044 to 0.0294; figure 2). No systemic AESIs occurred in the bilaterally treated patients during the 15-day and 30-day risk periods, so it was not possible to calculate the AIR for this subpopulation. This could be due to low probability of the events and the small size of the bilaterally treated patient population. In the self-controlled case series, with a 15-day and a 30-day risk period, the AIR was 21.6 cases per 1000 person-years (95% CI 0.0103 to 0.0330) and 24.1 cases per 1000 person-years (95% CI 0.099 to 0.0383), respectively (figure 2).
The self-controlled case series analyses considering a 15-day risk period and a 30-day risk period provided incidence rate ratios of 0.8253 and 0.6996, respectively, indicating a lower rate of AESIs in the risk period than in the control period.
The AIR of serious and non-serious systemic drug-related AEs and AESIs considering a 15-day and 30-day risk period are shown in table 3.
Annual incidence rates of serious and non-serious systemic drug-related AEs and AEs of special interest (Safety set*)
Annual incidence rate of ocular drug-related AEs and ocular AEs of special interest
There were nine ocular drug-related AEs reported during the study (table 4); one (retinal haemorrhage) was serious. Ocular AESI were reported in 11 eyes (1.1%); 6 (0.6%) events were related to ocular hypertension, 4 (0.4%) to intraocular inflammation and 1 (0.1%) to retinal tear (table 4); one event of ocular hypertension was serious but was considered by the investigator to be not related to treatment and the patient was retreated again.
Ocular drug-related AEs (by Preferred Term) and ocular AEs of special interest during the study (by Safety Risk and Preferred Term; Treated eyes of safety set patients*)
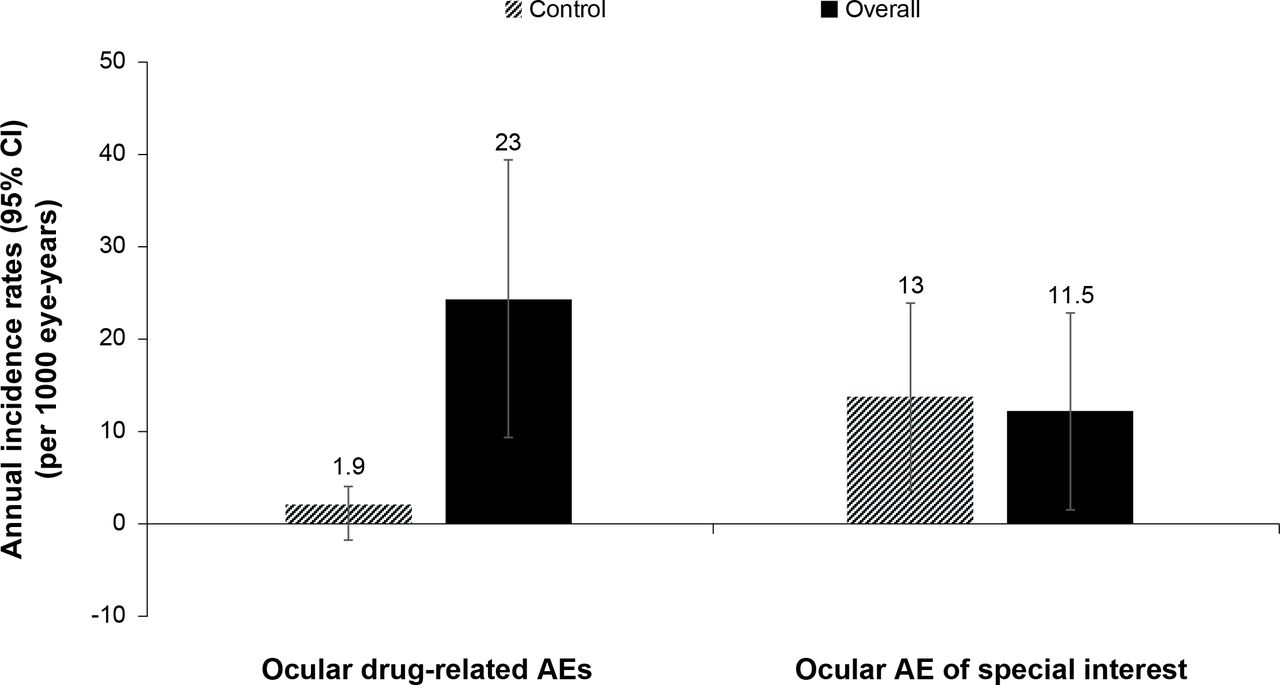
Using a 30-day risk period, there were 10 ocular drug-related AEs (of which one was serious) and five ocular AESIs (of which one was serious). The AIR of ocular drug-related AEs and ocular AESI over a 30-day risk period for the overall population and self-controlled case series are shown in figure 3. The number of events and AIR of drug-related AEs and AESI for the ocular safety subpopulation in sensitivity analyses were comparable with that of the safety population.
{kind=link}
{kind=link}
{kind=link}
Annual incidence rates of ocular drug-related AEs and AESIs—30-day risk period* (Safety set†). *Time at risk was defined as the sum of the 30 days following each ranibizumab injection. If a treated eye was given more than one injection, its risk period was defined as: (1) Time elapsed from the injection to the 30 subsequent days—for injections given more than 31 days from the previous; (2) Time elapsed from the first injection to the 30 days following the last injection—for injections given up to 31 days from the previous. †Consisted of all enrolled patients who signed the informed consent, received at least one dose of study drug and had at least one postbaseline safety assessment. The overall annual incidence rate of ocular drug-related AEs and AESIs was derived as the total number of ocular drug-related events that occurred during the risk period divided by the treated eye-time-at-risk. Treated eye-time-at-risk in years was defined as time elapsed, for any ranibizumab injection, from the injection to a maximum of 30+1 subsequent days. Lower limits less than 0 were set to 0. AE, adverse event; AESI, adverse event of special interest.
The incidence rate ratio, risk versus control, was 0.8848, indicating a rate of events during the risk period practically equal to that of the control period.
The AIR of serious and non-serious ocular drug-related AEs and AESIs were low (table 5).
Annual incidence rates of serious and non-serious ocular drug-related AEs and ocular AEs of special interest using a 30 day risk period (Treated eyes of safety patients*)
Proportion of ocular and systemic adverse events
Of the 1872 eyes of the safety set patients (both eyes of each patient-treated and not treated), 127 eyes (6.8%) had at least one ocular AE after the first drug injection for a total of 167 ocular events (table 6). The most commonly reported ocular AEs were conjunctivitis (0.8%), cataract (0.7%), conjunctival haemorrhage (0.7%), blepharitis (0.4%) and retinal epithelium detachment (0.3%; table 6). Ocular serious adverse events (SAEs) were reported in three eyes (table 6).
Ocular adverse events (Safety set*)
A total of 255 systemic drug-related AEs in 157 patients in the safety set were reported during the study, 209 AEs in 132 unilaterally treated patients and 46 AEs in 25 bilaterally treated patients. Systemic SAEs were reported in 60 (6.4%) patients, 51 (5.8%) unilaterally treated and 9 (8.0%) bilaterally treated (table 7).
Systemic adverse events (Safety set*)
Ocular AEs in two eyes resulted in treatment discontinuation (retinal pigment epithelium detachment and retinal degeneration) which was considered by the investigator to be treatment-related. Systemic AEs in 15 patients resulted in treatment discontinuation. There were nine deaths reported during the study (pulmonary cancer, sepsis, heart attack, septicaemia, pulmonary embolism, myocardial ischaemia, cardiac failure [all n=1 each], lung cancer [n=2]), but none of them were considered by the investigator to be related to the study drug.
Discussion
The TWEYEs study is the first real-life pragmatic study in Italy that assessed the safety and tolerability of ranibizumab 0.5 mg in patients with unilateral or bilateral nAMD and with low VA of <2/10. Ranibizumab treatment was well-tolerated in patients with nAMD. The population included in this trial was of high risk considering that they had a high presence of comorbidities and more than 50% of the patients were 75–84 years of age.
Anti-VEGF agents have revolutionised the treatment of visual impairment in patients with nAMD.24 25 Clinical experience in oncology has shown that systemic VEGF inhibition is associated with several ‘classes’ of AEs.26 Although the safety of intravitreal anti-VEGF agents has been studied extensively, a full understanding of the risk of ocular and/or systemic AEs possibly related to VEGF inhibition after intravitreal administration, especially in high-risk population is still lacking owing to paucity of data.25 Also, in Italy, the AIFA registry did not routinely capture data for patients with nAMD who had bilateral treatment or had low baseline VA.
Ranibizumab, an anti-VEGF agent specifically designed for intravitreal administration, has a short intrinsic systemic elimination half-life (2 hours),23 and low serum concentrations (below the concentration necessary to inhibit the biological activity of VEGF by 50%) following intravitreal injection in patients with nAMD. Subsequently ranibizumab does not largely affect plasma VEGF or free VEGF levels after intravitreal administration.27–29
The TWEYEs study assessed systemic and ocular AESIs (which included all AEs possibly related to ranibizumab as per the PSUR and RMP effective at the time of the study), in addition to AEs with causal relationship established by the investigator, in order to provide the most comprehensive and objective ranibizumab-related AE profile. The AIR of systemic AESIs was 17.8 events per 1000 person-years considering a 15-day risk period following each ranibizumab injection and 16.9 events per 1000 person-years considering a 30-day risk period. Moreover, in TWEYEs, treating patients in both eyes did not increase the likelihood of systemic events, as systemic AESIs were observed only in two bilaterally treated patients, both events occurred outside of the risk periods. These findings are in line with those reported previously.30 31
Of the known AEs associated with systemic VEGF inhibition, the risk of arterial thromboembolic events are of particular concern. The incidence of stroke/non-myocardial arterial thromboembolic events (0.5% [n=5] including cerebral haemorrhage, cerebrovascular accident and TIA) reported in this study was similar to what was observed in the pooled analysis of four European registries (0.4%).22
Consistent with findings from randomised controlled trials (ANCHOR, MARINA and SUSTAIN) and the analysis of four European registries,14 15 22 32 there was a low number of ocular events with ranibizumab injections, which might have been caused by the intravitreal injections procedure or by the anti-VEGF effect. The AIR of ocular AESI occurring within a 30-day risk period was 11.5 events per 1000 eye-years and consisted of IOP increase, intraocular inflammation and one case of retinal tear. Overall, the safety findings from this study were comparable to other pivotal clinical trials with no new critical safety findings.14 15 22 32 The mean age of patients enrolled in TWEYEs was similar to those enrolled in ANCHOR, MARINA and SUSTAIN studies (mean age of approximately 76–79 years).14 15 32 However, comparison across studies should be interpreted with caution considering the difference in study design, treatment patterns and eligibility criteria.
The mean number of injections per patient of the safety population over 1 year in this trial (6.0) is comparable to the mean numbers reported in the observational LUMINOUS study (5.0 injections)22 and that observed in EPICOHORT (4.4 in the first year), a 2-year,multicentre, open-label, observational, non-comparative study on patients with nAMD from 54 European clinical centres.31 These mean numbers are similar to the mean number of injections (5.6) reported in SUSTAIN, a randomised trial with PRN dosing.32 In a multicentre, nAMD database study conducted in the UK involving 12 951 eyes of 11 135 patients, the mean number of injections in the first year for the first treated eyes was 5.6.16 Observations made in TWEYES study in real-life setting in Italy showed that in actual clinical practice, the treatment pattern does not necessarily follow the recommended treatment regimen, in spite of treatment being available free of charge. Not all patients received at least three initial injections as recommended in the label, and the proportion of patients receiving injections decreased steadily starting from month 4. Possible reasons for this could be the busy schedules in clinics leading to inadequate patient monitoring and treatment, and the disease not being considered as an emergency condition like, for example, cardiovascular disease. Also, it was observed that many patients who did not meet study entry criteria were included in this study and more than 50% patients were not monitored monthly for VA.
The limitation of the study is that only about one fifth of the planned eyes (target 5000 eyes) were enrolled due to a lower than expected enrolment rate. As a result, the CI for the AIR of ocular drug-related AEs was greater (2.85%) than what was planned (0.6%). In addition, there was no systematic gathering of data on prior anti-VEGF medications and this could have led to bias in some patients treated with other medications previously. In addition, previous data related to systemic anti-VEGF therapy used for cancer treatment were not collected systematically leading to a possible bias in this subpopulation. The inability to collect systemic AEs in a systematic manner should also be taken into consideration and this is reflective of the considerable differences observed between drug-related AEs, as judged by the investigators, and AIRs of AESI.
The strength of the study is that it is one of the first studies to enrol a large number of patients for evaluating the safety of ranibizumab 0.5 mg in nAMD, especially in those with VA <2/10 and those with bilateral nAMD treatment. The pragmatic study design, method of data collection and statistical analysis were such as to ensure both the study’s internal validity and the applicability of the results to other settings, samples and populations.
In conclusion, the TWEYEs study results confirm the good safety and tolerability profile of ranibizumab 0.5 mg, in both unilaterally and bilaterally treated, mostly elderly patients with nAMD over a 1-year period and in a pragmatic real-world setting characterised by high number of protocol deviations and not all patients were treated as per label. The low incidence of systemic events in this pragmatic clinical study of a predominantly elderly population appear to be consistent with the known low systemic exposure and short half-life of ranibizumab.
Acknowledgments
The authors thank Sabyasachi Ghosh and Lakshmi Venkatraman (Scientific Services Practice – Product Lifecycle Services, Novartis Healthcare Pvt. Ltd., Hyderabad, India) for medical writing and editorial assistance towards the development of this article. The TWEYEs study authors would like to acknowledge all principal investigators across all sites who contributed to this study (in alphabetical order): Acquaviva Antonio, Addabbo Giuseppe, Anfossi Roberto, Arvedi Paolo, Avitabile Teresio, Azzolini Claudio, Belloli Vito, Beltrame Giorgio, Bocca Lidia, Bordin Paolo, Borin Stefano, Boscia Francesco, Cagini Carlo, Campos Emilio, Cappuccini Luca, Cataldo Stefano, Cau Antonello, Ciardella Antonio, Chizzolini Marzio, Cillino Salvatore, Cipollone Ugo, Conti Massimo, Coser Stefano, Crugliano Gennaro, De Castro Silvia, De Cillà Stefano, De Crecchio Giuseppe, De Rosa Pasquale, Dolcino Daniela, Eandi Chiara Maria, Ermini Dario, Falcomatà Bruno, Fantaguzzi Paolo, Fava Sebastiano, Fedeli Romolo, Figini Innocente, Fioretto Mauro, Fossarello Maurizio, Galan Alessandro, Gallo Otello, Gambaro Stefano, Gandolfi Stefano, Gasparri Vito, Giovannini Alfonso, Giustolisi Rosalia, Greco Alfredo, Gori Simona Giuseppina, Iaculli Cristiana, Iovieno Giovanni, Kacerik Miroslav, Laborante Antonio, Lanzetta Paolo, Lavezzari Paolo, Lopresti Giovanni, Lupidi Giovanni, Murialdo Ugo, Marchini Giorgio, Marino Antonio, Marullo Michele, Mastropasqua Leonardo, Minnella Angelo, Micelli Ferrari Tommaso, Menga Massimo, Macinagrossa Giancarlo, Nucci Paolo, Pece Alfredo, Perri Paolo, Pertile Grazia, Pezzato Paolo, Pioppo Antonino, Pirozzi Enza, Prosdocimo Giovanni, Prosio Pier Elio, Polvicino Mario, Prantera Marcello, Pucci Vincenzo, Romano Ferdinando, Rossetti Luca, Rossi Marco, Sbordone Giovan Battista, Sbordone Mario, Scarpa Giuseppe, Sciacca Riccardo, Scorcia Vincenzo, Scudieri Gianluca, Semeraro Francesco, Simonelli Francesca, Sperti Francesco, Surace Dario,Tognetto Daniele, Trillo Carlandrea, Traverso Carlo Enrico, Trabucchi Giuseppe, Trombetta Costantino, Troiano Antonello, Varano Monica, Longo Salvatore, Vingolo Enzo Maria, Vandelli Giulio, Vinciguerra Paolo, Vittici Vincenzo, Zotti Carlo Alberto.
References
Footnotes
Correction notice This paper has been amended since it was published Online First. The TWEYEs study authors have added an acknowledgement to the principal investigators across all the sites who contributed to this study.
Contributors Conception and design: FB, GS, EP. Analysis and interpretation: FB, GS, FR, EM, FV, TLS, LC, EP, SB. Data collection: TLS, LC, EP, SB. Overall responsibility: FB, GS, FR, EM, FV, TLS, LC, EP, SB.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests FB: Consultant—Acucela (Seattle, Washington), Alcon (Fort Worth, Texas), Bayer (Leverkusen, Germany), Boehringer-Ingelheim (Ingelheim, Germany), Genentech (South San Francisco, California), Heidelberg Engineering (Heidelberg, Germany), Novartis (Basel, Switzerland), Roche (Basel, Switzerland) and Zeiss (Oberkochen, Germany); Financial support—Bayer (Leverkusen, Germany), Genentech (South San Francisco, California), Heidelberg Engineering (Heidelberg, Germany), Novartis (Basel, Switzerland), Roche (Basel, Switzerland) and Zeiss (Oberkochen, Germany). GS: Consultant—Novartis (Basel, Switzerland), Bayer HealthCare (Leverkusen, Germany), Allergan (Dublin, Ireland), Genentech (South San Francisco, California), Roche (Basel, Switzerland), Heidelberg Engineering (Heidelberg, Germany) and Alcon (Fort Worth, Texas); Financial support—Bayer HealthCare (Leverkusen, Germany), Centervue (Padova, Italy), Heidelberg Engineering (Heidelberg, Germany) and Novartis (Basel, Switzerland); Lecturer—Zeiss (Oberkochen, Germany); Patent holder—in conjunction with Ocular Instruments, Inc. (Bellevue, Washington); Payment for development of educational presentations—Roche (Basel, Switzerland). FR: Alcon (Fort Worth, Texas), Bayer (Leverkusen, Germany), Novartis (Basel, Switzerland), Roche (Basel, Switzerland), Allergan (Dublin, Ireland), SIFI (Catania, Italy). FV: Consultant—Novartis (Basel, Switzerland), Bayer HealthCare (Leverkusen, Germany), Financial support—Allergan (Dublin, Ireland), Novartis (Basel, Switzerland), Bayer HealthCare (Leverkusen, Germany), SIFI (Catania, Italy). TLS: Employee—Novartis Farma S.p.A (Origgio, Italy) at the time of development of the manuscript and submission. EP, SB: Employees—Novartis Farma S.p.A (Origgio, Italy). LC: Employee—Novartis Farma S.p.A (Origgio, Italy) at the time of development of the manuscript and submission.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on request.
Linked Articles
- At a glance