Article Text

Abstract
Aims To identify the mutation spectrum and genotype–phenotype correlations of fibrillin-1 (FBN1) mutations in a Chinese cohort with congenital ectopia lentis (EL).
Methods Patients clinically suspected of congenital zonulopathy were screened using panel-based next-generation sequencing followed by multiplex ligation-dependent probe amplification. All the probands were subjected to thorough ocular examinations. Molecular and clinical data were integrated in pursuit of genotype–phenotype correlation.
Results A total of 131 probands of FBN1 mutations from unrelated families were recruited. Around 65% of the probands were children younger than 9 years old. Overall, 110 distinct FBN1 mutations were identified, including 39 novel ones. The most at-risk regions were exons 13, 2, 6, 15, 24 and 33 in descending order of mutation frequency. The most prevalent mutation was c.184C>T (seven, 5.34%) in the coding sequence and c.5788+5G>A (three, 2.29%) in introns. Missense mutations were the most frequent type (103, 78.63%); half of which were distributed in the N-terminal regions (53, 51.46%). The majority of missense mutations were detected in one of the calcium-binding epidermal growth factor-like domains (62, 60.19%), and 39 (62.90%) of them were substitutions of conserved cysteine residues. Microspherophakia (MSP) was found in 15 patients (11.45%). Mutations in the middle region (exons 22–42), especially exon 26, had higher risks of combined MSP (OR, 5.51 (95% CI 1.364 to 22.274), p=0.017).
Conclusions This study extended the knowledge of the FBN1 mutation spectrum and provided novel insights into its clinical correlation regarding EL and MSP in the Chinese population.
- genetics
- lens and zonules
Data availability statement
Data are available in a public, open-access repository. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The fibrillin-1 (FBN1) gene encodes a 350 kDa cysteine-rich glycoprotein that serves as the principal structural component of microfibrils, which contribute to the force-bearing capacity of connective tissue such as blood vessels, ligaments and bones. More than 3000 mutations of FBN1 are known today,1 which are associated with a broad spectrum of phenotypes, collectively named ‘type I fibrillinopathy’. Marfan syndrome is the most well-known one and is typically characterised by aortic dilation, lens dislocation and the overgrowth of tubular bones.2 Other isolated features and systematic involvement have been observed in FBN1 mutation carriers, including familial ectopia lentis (EL); isolated ascending aortic aneurysm; stiff skin syndrome; mitral valve prolapse syndrome, mitral valve prolapses, aortic dilatation, and skeletal and skin involvement syndrome; autosomal dominant Weill-Marchesani syndrome; and Shprintzen-Goldberg syndrome.3
EL is the dislocation of the lens from its anatomical position, which is one of the cardinal features when diagnosing Marfan syndrome.4 The dislocation is usually progressive and bilateral and has a superior direction.5 In addition to EL, FBN1 mutations can lead to various kinds of zonular abnormalities, which are termed ‘zonulopathy’: segmental zonular compromise may result in coloboma lentis, while global weakness can lead to microspherophakia (MSP). The degree of zonular weakness is also variable: a dislocated lens with a round edge suggests the residual zonules are insufficiently strong, whereas an irregular edge indicates the presence of relatively strong fibres on the protruding sites.6 The mechanisms underlying the diverse manifestations are largely unknown but are likely to be due to the different types or sites of FBN1 mutations. In this study, we investigated the spectrum of FBN1 mutations using panel-based next-generation sequencing (NGS) followed by multiplex ligation-dependent probe amplification (MLPA). The purpose was to identify the at-risk regions and hotspots for FBN1 mutations in a Chinese cohort of congenital zonulopathy (mainly EL). We also intended to investigate the possible correlations between MSP and FBN1 mutation. The results will potentially expand our understanding of FBN1-associated EL, MSP and related disorders.
Methods
Patients eligibility
The inclusion criteria in our study were (1) the presence of a heterozygous pathogenic or likely pathogenic FBN1 mutation; (2) a diagnosis of EL, MSP or coloboma lentis under slit-lamp examination during complete pupillary dilation; and (3) the availability of clinical information. Patients with the following features were excluded from the study: (1) a history of traumatic EL and (2) the coexistence of pathogenic mutations of other ocular diseases. To avoid selection bias as a result of familial clustering, only the probands of pedigrees were enrolled. The age at the first observation of clinical features was recorded.
Ophthalmic examinations
The participants were examined by board-certified ophthalmologists, and their medical and family histories were evaluated. All patients received a slit-lamp examination under complete pupillary dilation. EL was defined as the displacement of the lens from its normal position (figure 1A). MSP was diagnosed by increased lens thickness and reduced equatorial diameter (figure 1B). Coloboma lentis was diagnosed when there was a notch in the lens tissue at the equator (figure 1C). One example of a dislocated lens with an irregular edge was presented (figure 1D). Posterior staphyloma was examined by B-scan ultrasound. Ciliary body cyst was detected by ultrasound biomicroscopy (BMD-300 L, MEDA, Tianjin, China). The intraocular pressure was measured with a non-contact tonometer (CT-80, Topcon, Oakland, USA).

Photos of congenital zonulopathy caused by fibrillin-1 mutations. (A) One eye of a patient (c.1709G>C) with EL and smooth lens edge. The lens was dislocated superior nasally. (B) One eye of a patient (c.3476G>T) with EL and MSP. The lens was thicker, and the entire equator was visible when the pupil was fully dilated. The superior equator is hidden because of combined EL. (C) One eye of a patient (c.290C>G) with coloboma lentis. A notch in the lens tissue at the equator was observed inferior temporally. (D) One eye of a patient (c.5885_5895del) with EL and an irregular lens edge at the temporal side. EL, ectopia lentis; MSP, microspherophakia.
Genetic screening
Genomic DNA was extracted from peripheral blood using the CWE2100 Blood DNA Kit (CW2526S, CWBIO, Cambridge, Massachusetts, USA). We employed the panel-based NGS produced by Amplicon Gene (Shanghai, China), which was custom-designed to include the exon sequences of 289 genes involved in common inherited anterior eye diseases, with the extension of the exon area by 30 bp on either side (online supplemental table 1). The promoter regions, flanking intronic regions and coding exons were captured using the Dynabeads MyOne Streptavidin T1 (65601, Invitrogen, Waltham, Massachusetts, USA). The enriched libraries were sequenced on Illumina NovaSeq 6000 (Illumina, San Diego, California, USA). Sequencing reads were mapped to the reference human genome (UCSC hg19) using a Burrows-Wheeler Aligner (http://bio-bwa.sourceforge.net/). The frequencies of identified variants were annotated through the 1000 Genomes Project (http://browser.1000genomes.org). Sanger sequencing was performed to confirm the candidate mutations of the patients and their family members. For the MLPA assay, the commercially available SALSA MLPA Probemix kits (MRC Holland, number P065-C1/P066-C1) were used. MLPA was performed using 100 ng of DNA in a 5 mL reaction. MLPA-generated fragments were detected using a capillary electrophoresis system (ABI3730XL, Applied Biosystems, Waltham, Massachusetts, USA), and we confirmed the positive MLPA results using quantitative PCR.
Supplemental material
Genetic analysis
The pathogenic nature of each FBN1 mutation was evaluated using Ghent-2 criteria and was classified as causal or unknown.4 Pathogenicity was also predicted using Polymorphism Phenotyping V.2 (http://genetics.bwh.harvard.edu/pph2/) and MutationTaster software (http://www.mutationtaster.org/). Splicing site mutations were identified using Human Splicing Finder (http://www.umd.be/HSF/) and SPANR (http://tools.genes.toronto.edu/). All mutations were classified as pathogenic, likely pathogenic, uncertain clinical significance and likely benign under the American College of Medical Genetics and genomics guidelines.7 All mutations were reviewed using the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), Human Gene Mutation Database (www.hgmd.cf.ac.uk/ac/) and Universal Mutation Database of FBN1 (http://umd.be/FBN1/).8 Divisions of the gene into the N-terminal region (exons 1–21), C-terminal region (exons 43–65) and midregion (exons 22–42) were made according to a previous study.9
Statistical analyses
Student’s t test or Mann-Whitney U test was applied to compare the continuous variables of cohort characteristics between two groups. The one-way analysis of variance or Kruskal-Wallis test was used to compare patient ages in the different genotype groups. χ2 test, Yates’ correction or Fisher’s exact test was employed to compare the mutation spectrum of mutations. The statistical significance was set as a P value of <0.05, unless otherwise specified. Statistical analyses were performed using SPSS V.25.0. The cluster method was Ward.D2 using R, V.4.0.1 (R Foundation for Statistical Computing).
Results
Cohort characteristics
A total of 158 probands of congenital zonulopathy received a panel-based NGS test in Fudan University Eye and ENT Hospital between 2016 and 2020. The mean age was 11.45±11.38 years old. Pathogenic or likely pathogenic FBN1 mutations were detected in 129 probands. Fifteen probands had mutations in other genes. In 14 probands without the pathogenic genes, four patients highly suspected of Marfan syndrome underwent the MLPA assay. One allele deletion of FBN1 and one intragenic duplication of exon 34 (g.(171595_173174)_(173299_175094)dup) were detected in two patients. Thus, a total of 110 mutations of 131 probands (including 11 recurrent mutations in 32 probands) were enrolled in this study (online supplemental table 2). The segregation data are shown in online supplemental table 3. The demographical statistics are summarised in table 1. All but one patient had EL. One 18-year-old patient (c.290C>G) had bilateral coloboma lentis on the temporal-inferior side without evidence of EL. MSP was complicated in approximately 11% of patients, and a lens with an irregular edge was observed, along with EL, in around 14% of patients. More than half of the patients were the only affected members of their families. Most of the patients were diagnosed with potential Marfan syndrome, followed by Marfan syndrome and EL syndrome.
Supplemental material
Supplemental material
Demographical status of the 131 probands with fibrillin-1 mutations
Genetic spectrum
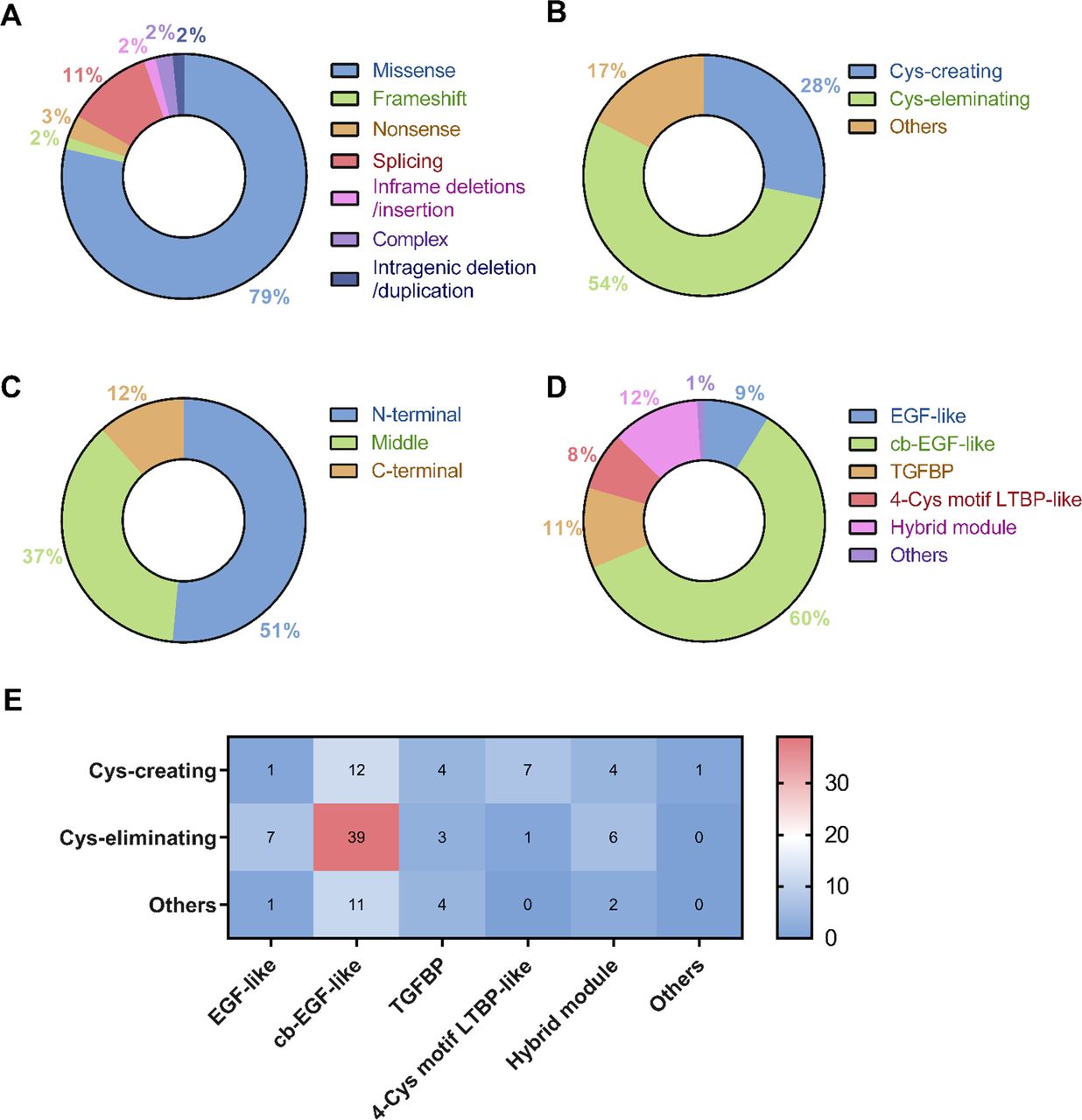
Mutation distributions (recurrent ones included) were identified in 40 of 65 exons and 11 of 65 introns. The regions harbouring the most mutations and, thus, considered most at risk of mutation were exons 13, 2, 6, 15, 24 and 33 in descending order of frequency (figure 2A). One silent mutation (c.6354C>T, p.Ile2118Ile) was mapped on exon 51, and it was reported to induce in-frame skipping of the entirety of exon 51. There were 14 mutations observed in 11 introns (figure 2B). Three probands harboured c.5788+5G>A, which was the most prevalent splicing mutation (2.29%), and have been demonstrated to cause exon 46 skipping.10 However, other splicing mutations had unclear mutation causality. The most prevalent mutation in the coding sequence was c.184C>T (seven, 5.34%), followed by c.4096G>A (four, 3.05%) and c.718C>T (four, 3.05%) (figure 2C). The proportions of mutation types and locations are summarised in figure 3. The majority of probands carried missense mutations (103, 78.63%); the amino acid change of which mainly involved cysteine (85, 82.52%) (figure 3A,B). The N-terminal had the most missense mutations (53, 51.46%) (figure 3). Of the nine functional domains of the FBN1 protein, the calcium-binding (cb) EGF-like domain contained the majority of the missense mutations (62, 60.19%) (figure 3D). When taking both amino acid changes and locations into consideration, the largest proportion (39, 37.86%) of missense mutations involving conserved cysteine residues occurred in the cb-EGF-like domain (figure 3E).

Structural distribution of fibrillin-1 (FBN1) mutations (including recurrent mutations). (A) Frequency of FBN1 mutations per exon and the corresponding N-terminal (exons 1–21), middle (exons 22–42) and C-terminal (exons 43–65) regions. (B) Frequency of FBN1 mutations per intron. (C) Frequency of FBN1 mutations per amino acid and the corresponding protein domains. Hotspots are noted.cb, calcium binding; COOH; carboxyl; EGF, epidermal growth factor; LTBP, latent transforming growth factor β binding protein; NH2, amino; TGFBP, transforming growth factor β binding protein.

Genetic analyses of the fibrillin-1 (FBN1) mutations identified in 131 probands with zonulopathy. (A) Proportions of FBN1 mutations of all probands classified as missense mutations, frameshift mutations, nonsense mutations, splicing mutations, in-frame deletions or insertions, complex mutations and intragenic deletion/duplication. (B) Proportions of FBN1 missense mutation amino acid changes classified as cysteine (Cys)-creating, Cys-eliminating and changes involving other amino acids. (C) Proportions of FBN1 missense mutations mapped to coding sequences in the N-terminal (exons 1–21), middle (exons 22–42) and C-terminal (exons 43–65) regions. (D) Proportions of FBN1 missense mutations mapped to EGF-like domain, cb-EGF-like domain, TGFBP domain, 4-Cys motif LTBP-like domain, hybrid module and other protein domains. (E) Heatmap showing the frequency of Cys-creating, Cys-eliminating and other types of amino acid changes in different protein domains. cb, calcium binding; EGF, epidermal growth factor; LTBP, latent transforming growth factor β binding protein; TGFBP, transforming growth factor β binding protein.
Database analysis
Among the 110 mutations found in the 131 probands, 39 mutations from 40 probands were reported for the first time, including 28 missense mutations, one frameshift mutation, four splicing mutations, one in-frame deletion, one allele deletion and one intragenic duplication. Three mutations involving more than one type of mutation were categorised as complex mutations (c.(355T>C) + (3142A>G), c.(1884C>G) + (7241G>A) and c.4817–42_4817-7del). There are 29 mutations from 31 probands without phenotype information in online databases. The phenotypes of 42 mutations from 60 probands were reported in the literature (online supplemental table 2).
Genotype–phenotype correlations
Since the age of onset was unknown for most of the patients, we investigated the relationship between age at the first clinical observation of EL and genotype. However, there were no differences in genotype among patients diagnosed at different ages (online supplemental figure 1). Fifteen probands were diagnosed with MSP. The cohort characteristics were similar between groups with and without MSP (online supplemental table 4). The only genotype–phenotype correlation was found in terms of mutation sites: missense mutations in middle regions (exons 22–42) had a higher risk of developing MSP than the other regions (OR, 5.51 (95% CI 1.364 to 22.274), p=0.017). (figure 4A). With regard to individual exons, exon 26 was the location of three mutations (c.3217G>A, 3229T>C and C.3244G>T) from four probands diagnosed as having MSP, which had a significantly higher prevalence than the other exons (Fisher’s exact test, α=0.05/65, p=7.5×10−5) (figure 4B). No correlations were observed for other exons or introns (data not shown). Moreover, there were no correlations between mutation types and MSP (Fisher’s exact test, p=0.283). For missense mutations, there were no correlations between MSP and amino acid change (Fisher’s exact test, p=0.760) or domain distribution (Fisher’s exact test, p=0.669) (figure 4A).
Supplemental material
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Genotype–phenotype analysis of patients with and without MSP. (A) Comparison of type of mutation, amino acid change, protein domain and mutation sites in patients with EL alone or EL complicated by MSP. (B) Comparison of mutation spectrum by cluster analysis in patients with EL alone or EL complicated by MSP. Heatmap of fibrillin-1 mutations shows the number of missense mutations per exons and splicing mutations per introns. Exon 26 is marked by a red asterisk. Cys, cysteine; EL, ectopia lentis; EL − MSP, patients with EL but not MSP; EL+ MSP, patients with EL and MSP; EX, exon; Cys, MSP, microspherophakia. cb, calcium binding; EGF, epidermal growth factor; LTBP, latent transforming growth factor β binding protein; TGFBP, transforming growth factor β binding protein.
Discussion
FBN1 comprise 64% of Zinn’s zonule, which suspends the lens in the optical centre and transmits tension from the ciliary body in disaccommodation.11 Marfan syndrome and FBN1 mutations are the main causes of congenital EL,12 13 which is ranked the second reason for lens surgery in childhood after congenital cataracts.14 In the present study, we characterised the mutation spectrum of FBN1 based on phenotype selection. We identified 110 EL-associated FBN1 mutations in 131 probands by NGS followed by MLPA, and 39 of the mutations were previously unidentified.
More than 3000 mutations of FBN1 have been reported.8 In the present study, the most commonly observed mutations were missense; the majority of which involved cysteine. The regions with the highest risk of mutation in this study were exons 13, 2, 6, 15, 24 and 33, which agrees with previous findings in exons 2, 6 and 13.15 Missense mutations tended to cluster in the N-terminal regions, which has been shown to have a strong association with EL.15 The N-terminal region is responsible for the assembly of fibrillin and the stability of microfibrils.16 This region can also interact with latent TGF-β-binding protein 2 (LTBP2), a bridging structure among microfibrils.17 Thus, disruption of this region is likely to impair the tensile properties by weakening lateral interactions among microfibrils, which is consistent with the observation that the zonules may appear intact but were hyperextended in the eyes of Marfan syndrome.5 Of the various functional domains, cb-EGF-like domains were found to carry significantly more missense mutations, especially those eliminating cysteine. Cysteine substitutions can disrupt the disulfide bridges covalently connecting three pairs of cysteine residues.18 The misfolding of cb-EGF-like domains has been shown to arrest intracellular trafficking and diminish extracellular deposition.19 Thus, the conserved cysteine residues on the cb-EGF-like domains may have important roles in the structural integrity of microfibrils. The additional mutations in this domain probably disturb the calcium binding involved in resisting proteolytic degradation.20 Approximately 11% of patients had mutations affecting splicing sites, which were reported to lead to a high possibility of combined EL.21 Although little is known about the mutational products, the majority of splicing site mutations result in in-frame exon skipping and the loss of an integral domain.22 Further functional studies of the mRNA products of splicing events will help us to understand their clinical significance.
FBN1 mutations were thought to be unique to a pedigree and generally non-recurrent, as the recurrence rate in the database was 12%.22 Our study reported a relatively higher recurrence rate of 24.43%, probably because the phenotype selection was based on EL and thus more specific. Our study identified c.184C>T (R62C) to be a mutational hotspot (seven, 5.34%). In another cohort, mainly from southern China, c.184C>T was also found to be the most frequent mutation (four, 11.76%).12 This mutation, which is predicted to replace an arginine with a cysteine at the 4-cysteine motif LTBP-like domain, was first reported in a 31-year-old man with isolated EL without any systematic involvement and a 25-year-old man with bilateral EL but mild dilation of the aortic root (46 mm on echocardiography).23 Two large Chinese pedigrees with isolated EL caused by this mutation have also been reported; all family members had normal aortic measurements and no skeletal involvement.24 25 In our study, all probands with c.184C>T had early-onset bilateral EL but did not fulfil the criteria of the Ghent-2 nosology. Less frequent mutations were c.4096G>A (four, 3.05%) and c.718C>T (four, 3.05%), which were both reported in patients of isolated EL.26 27 In patients with pathogenic FBN1 mutations and EL, but lacking cardiac involvement, the diagnosis ‘EL syndrome’ is suggested if the patient is over 20 years old.4 Given the highly dispersed distribution, correction of an FBN1 mutation by base editing may not be practical for most patients; however, specific editing of the hotspots is promising in the future.28
Besides allelic heterogeneity, FBN1 mutations also produce a wide phenotypic spectrum. Intensive efforts have been made to explore the genotype–phenotype correlations of the major involvements, such as EL, aortic dilation and a combination of skeletal features.29 However, studies concerning FBN1 mutations and minor manifestations in the ocular system are scarce. In the current study, MSP was complicated in around 11% of patients; none of which could be diagnosed as Weill-Marchesani syndrome. MSP is a rare condition in which the lens assumes a more spherical shape and a smaller size.30 The undersized lens is not due to inherent growth defects but secondary to a lack of tension from the rudimentary zonules. A previous study shows that the lens epithelial cell proliferation was modulated by mechanical strain via yes-associated protein (YAP) signalling.31 Thus, the lens without sufficient stretching remains more typical of the fetal period and fails to develop a biconvex shape after birth.30 The observation of MSP in patients with Marfan syndrome has occasionally been reported, but why a subset of patients develop MSP remains unknown. We found that patients with mutations distributed in the middle region (exons 22–42) were more likely to develop MSP than those with mutations in other regions. Mutations at exon 26 (c.3217G>A, 3229T>C and C.3244G>T) were all associated with MSP. This exon encodes one of the cb-EGF-like domains, which is located on exons 24–32, and this region has been intensively studied, as it is associated with neonatal Marfan syndrome.29 In line with previous findings, patients in our cohort with c.3217G>A and c.3229T>C showed classic Marfan syndrome at a young age.32 However, the two probands with c.3244G>T were both diagnosed as isolated EL after 45 years of age, and both had bilateral EL, MSP, cataracts and open-angle glaucoma but no skeletal or cardiac involvement. We postulated that the four mutations on exon 26 probably share a similar pathway leading to MSP but have distinct mechanisms leading to skeletal or cardiac phenotypes. The diagnosis of MSP is challenging; a few of which can only be found intraoperatively. A modified capsular tension ring (MCTR) with double eyelets should be applied if the surgeon plans in-the-bag intraocular lens implantation33; however, phacoemulsification is challenged by the globular laxity of the zonules, and the MCTR tends to oversize the capsular bag. Our results suggest that the surgeons should be cautious of MSP and make sufficient preparations before the surgery if the patient harbours an FBN1 mutation of exons 22–42, especially exon 26. Functional studies are warranted to pinpoint the mechanisms of MSP formation.
The results in this study should not be assessed without considering its limitations. First, the study did not present a complete mutation spectrum of the population. Patients who were reluctant to receive the genetic test were not enrolled. Second, the age at onset could not be obtained in most patients because those with early-onset EL generally have few visual complaints. Thus, the genotype–phenotypes can only be analysed concerning the age at clinical observation, and no difference was observed. Third, the genotype–phenotype analysis was only performed for MSP. Further analysis should focus on other ocular manifestations of FBN1 mutations, such as coloboma lentis, axial myopia, corneal abnormalities and retinal diseases. Additionally, skeletal and cardiac involvements were not discussed in this study because most of our patients were children with developing features. It will be interesting to see how the phenotypes of these patients evolve in the long-term follow-up.
In conclusion, our study provides a picture of FBN1 mutations in Chinese patients of EL, and the genotype–phenotype correlations for MSP in patients of EL were investigated for the first time. We anticipate that our work will provide illuminating reading for both genetic counsellors and cataract surgeons.
Supplemental material
Data availability statement
Data are available in a public, open-access repository. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by the human research ethics committee of the Eye and ENT Hospital of Fudan University (ChiCTR2000039132) and was carried out following the Declaration of Helsinki. Informed consent was obtained from all patients and the patients’ guardians for those under 18 years old.
Acknowledgments
The authors thank Dr Tong-Hai Dou from Amplicon Gene for her help in genetics counselling. The authors thank also Jia-Hao Mao, Yu-Xiang Zhan, and Yan Kang from Amplicon Gene for their assistance in collecting blood samples.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
ZC and TC contributed equally.
Contributors ZC and TC conceived and designed the experiments. MZ, MD, JC, JZ and L-NL collected the clinical samples. YJ, MZ and MD performed clinical examinations on patients and clinical interpretation. YJ, ZC, TC and MZ drafted and revised the manuscript. All authors read and approved the manuscript.
Funding This study was supported by the Shanghai Science and Technology Commission (Scientific Innovation Action Plan, grant number 20Y11911000), the National Natural Science Foundation of China (grant number 82070943) and the National Natural Science Foundation of China (grant number 81770908). The sponsors played no role in the study design, data collection, data analysis, manuscript preparation nor the decision to submit the manuscript for publication.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Linked Articles
- Highlights from this issue