Article Text

Abstract
Introduction: The mitochondrial DNA A3243G point mutation is associated with a wide variety of systemic manifestations including a macular dystrophy. The characteristics of fundus autofluorescence (AF) in these patients are distinctive and have not been previously described.
Methods: A complete history and ophthalmic examination, including fundus photography and autofluorescence imaging, was performed on twelve probands harbouring the A3243G point mutation.
Results: Four patients had diabetes, 10/12 hearing loss, and 7/12 were visually symptomatic. A positive family history was present in 5/12. Fundus findings consisted of two primary phenotypes: discontinuous circumferentially oriented perifoveal atrophy (9/12) or an appearance consistent with pattern dystrophy (3/12). In both phenotypes pale deposits and pigment clumping were seen at the level of the retinal pigment epithelium, with occasional changes also noted outside the arcades and nasal to the optic nerve. Fundus AF imaging revealed decreased autofluorescence in areas of atrophy and increased AF of the pale subretinal deposits. In areas of the retina that appeared normal clinically, variable sized flecks of increased and decreased AF were present.
Conclusions: The mitochondrial DNA A3243G point mutation can result in disease with a variable presentation. Fundus autofluorescence reveals a recognisable phenotype in most cases that is different from other macular dystrophies.
Statistics from Altmetric.com
The single-point mutation of the mitochondrial DNA (mtDNA) at the 3243 position in the tRNALeu(UUR) gene leads to a wide variety of clinical manifestations. This mutation was originally described in association with Mitochondrial Encephalopathy, Lactic Acidosis and Stroke-like episodes (MELAS), and is now known to produce a variety of other clinical disorders, including maternally inherited diabetes and deafness (MIDD), cardiomyopathy, chronic progressive external ophthalmoplegia (CPEO), a pure myopathy, gastrointestinal dysmotility and renal failure.1–6 One explanation for this variability is thought to be the load of mutant mtDNA present in an individual, which varies both between individuals and from tissue to tissue within an individual.7 In general, a higher mutation load is associated with more severe disease. However, this generalisation does not always hold true, and it is possible that environmental factors and/or nuclear genetic influences may modulate disease manifestations.6 8 9
Ocular manifestations of mitochondrial diseases are well recognised, and in the original report by Reardon et al which revealed a new subtype of diabetes caused by a mtDNA mutation, three patients were found to have retinal pigmentary changes.2 10 However, there were no fundus photographs of the changes described in this initial report.2 In 1995, Massin et al described the association of a bilateral macular pattern dystrophy with MIDD and further characterised this in a subsequent publication in 1999.11 12 Since these reports, there have been several additional publications describing the macular dystrophy of MIDD and MELAS, both clinically and electrophysiologically.13–18
The use of the confocal scanning laser ophthalmoscope (cSLO) for imaging of macular diseases has been well described.19–25 In this study, we have characterised in detail the distinct fundus autofluorescence (AF) characteristics of the macular dystrophy associated with the A3243G mtDNA mutation.
METHODS
All patients diagnosed as having a macular dystrophy due to the A3243G mtDNA mutation in the Medical Retina Clinic at Moorfields Eye Hospital between 1995 and 2001 were included in the study. Twelve patients in total were evaluated. Patient demographics, referring diagnosis, presence or absence of visual symptoms, duration of visual symptoms, general medical history including presence or absence of diabetes and deafness, maternal history of diabetes and deafness, and a detailed family history, were obtained for each patient. Best-corrected Snellen visual acuity and slit-lamp biomicroscopy findings were recorded. Colour fundus photographs and fundus autofluorescence imaging were performed. Patients were tested for the A3243G mutation based on the characteristics of their ophthalmic examination, regardless of whether or not there was a personal or family history of diabetes or deafness. The diagnosis of the A3243G mtDNA mutation was established with DNA testing of peripheral blood samples using previously described techniques.26
Fundus autofluorescence imaging was performed with a confocal scanning laser ophthalmoscope (cSLO, Zeiss, Jena, Germany or HRA, Heidelberg, Germany) using previously published techniques.19 20 27 Autofluorescence images were compared with colour photographs. The AF images of the patients with the A3243G mutation were also compared with images of patients with other maculopathies, including Stargardt macular dystrophy, R172W peripherin macular dystrophy, bull’s eye macular dystrophy, geographic atrophy from age-related macular degeneration and pattern dystrophy.
Patients who did not have a history of diabetes were tested with fasting blood glucose, where possible, according to the recommendations of the American Diabetic Association.28 Renal function tests were also performed when possible due to the association of renal dysfunction with the A3243G mutation.29
RESULTS
Twelve patients were diagnosed as having the A3243G mutation between January 1995 and February 2001; in none had the diagnosis of a mitochondrial disorder been made prior to being seen by the authors. The characteristics of these patients at presentation are summarised in table 1.
Nine of the patients were female. The average age at presentation was 47 years (range 36 to 65) with an average age at diagnosis of 51 (range 36 to 71). In six of 12 patients, the reason for referral to a specialist clinic was an unspecified maculopathy consisting of a combination of retinal pigment epithelial changes and atrophy. Other diagnoses at referral were Usher syndrome variant, central areolar choroidal sclerosis, pattern dystrophy, macular degeneration and macular atrophy. Five of the 12 patients did not have visual symptoms at presentation and were found to have fundus abnormalities on routine eye examination. Of the patients who were initially asymptomatic, two patients became symptomatic during review. Three patients are still asymptomatic after follow-up, ranging from 1 to 6 years. Asymptomatic patients tended to be younger than the patients with symptoms, with a mean age of 43 compared with 51 years of age, although the number of patients in each group is too small for statistical significance to be established. Of the seven patients with symptoms, three reported difficulties due to paracentral scotoma, and three reported a general decrease in vision in one eye.
A history of hearing loss was common, with 10 of 12 patients having some degree of hearing impairment. Patient 1 had a cochlear implant, and four other patients were using hearing aids. Diabetes was present in only four patients, all of whom also had hearing loss.
A maternal history of diabetes was present in two patients and a maternal history of deafness in five patients. A maternal history of both diabetes and deafness was elicited in only one subject. There were two additional patients with a maternal history of hearing loss diagnosed after the age of 75 years. This was considered to be age-related hearing loss and not included as a positive history in the context of a mitochondrial disease. Two further patients had a family history of maternal diabetes, which was not diagnosed until the age of 60, and was again not considered as a positive finding in the context of the A3243G mutation.
In all patients, presenting visual acuity was good, each having at least 6/9 or better vision, and seven patients achieving 6/6 or better vision, with each eye. At final follow-up, the vision was slightly worse, with all patients having 6/12 or better vision in at least one eye. Six out of 12 patients had 6/6 or better vision with at least one eye at the last documented examination. Of the four patients with 6/12 or worse vision, three had deterioration in vision due to paramacular atrophy encroaching upon fixation, and one had decreased vision bilaterally due to retinal pigment epitheliopathy.
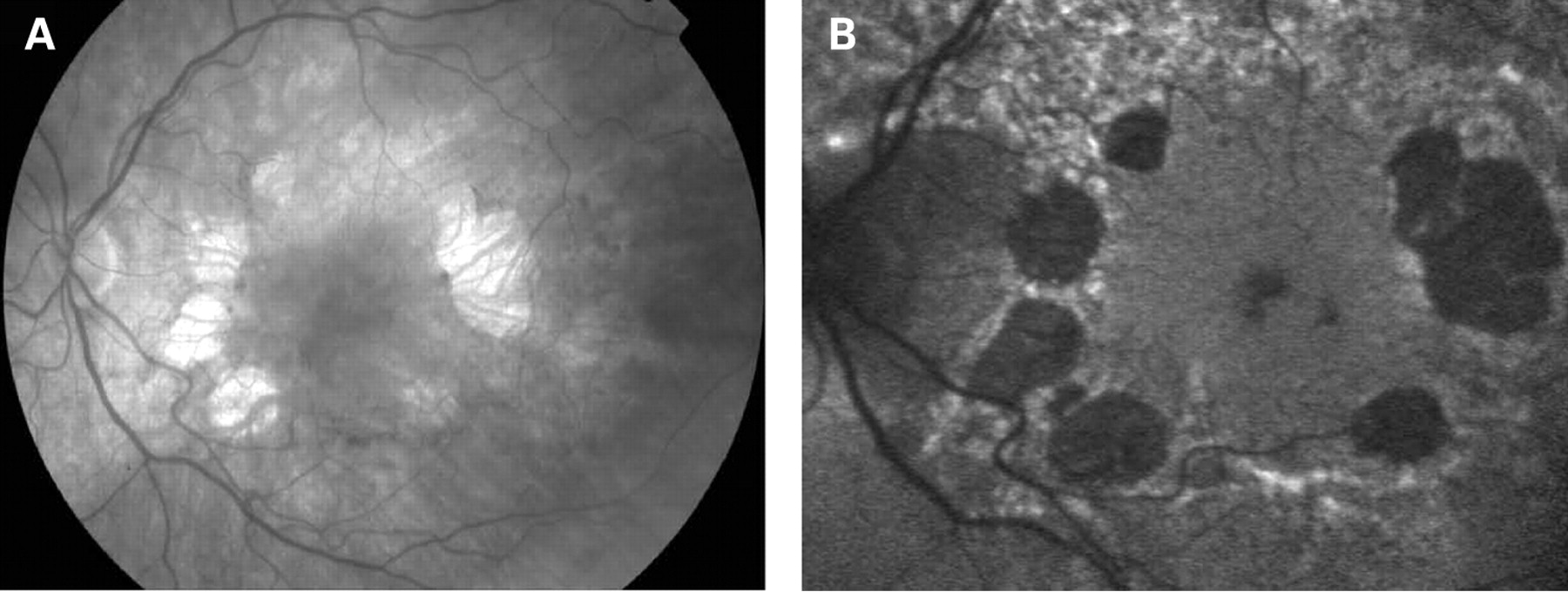
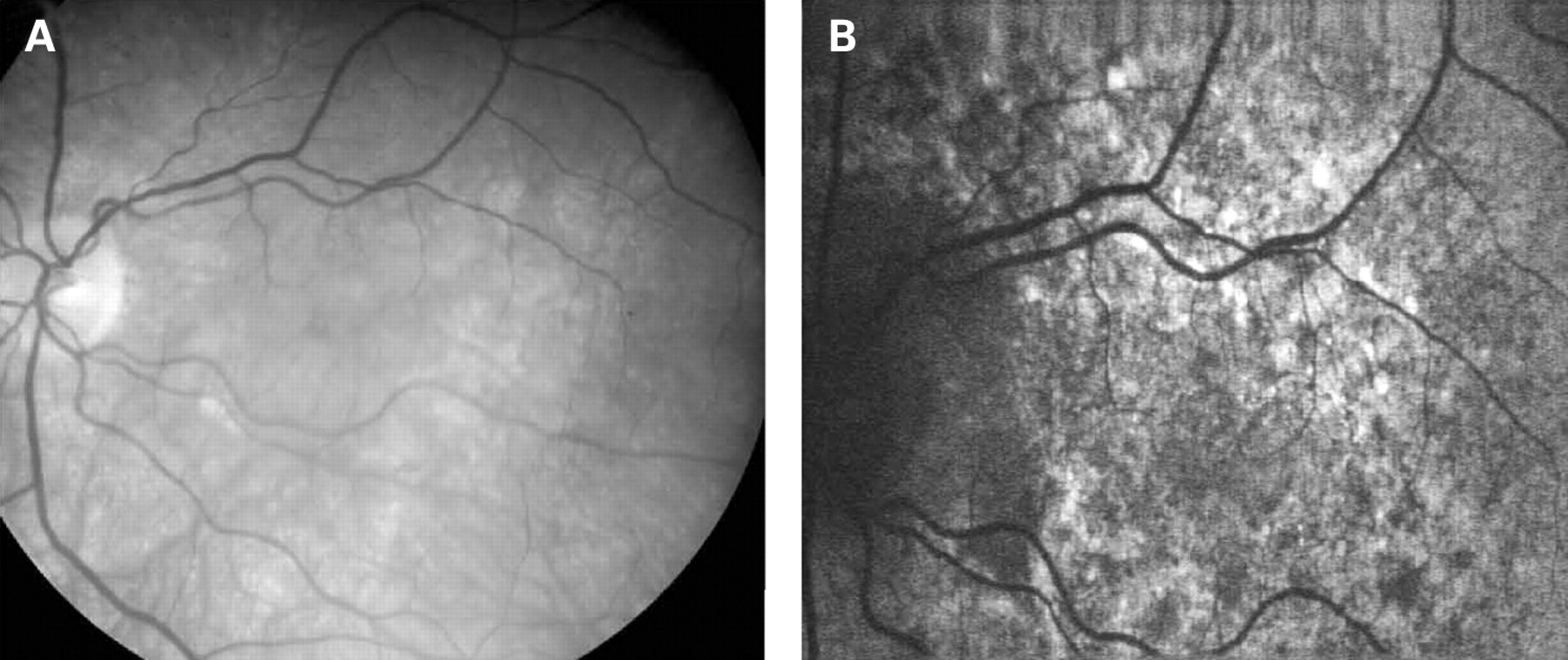
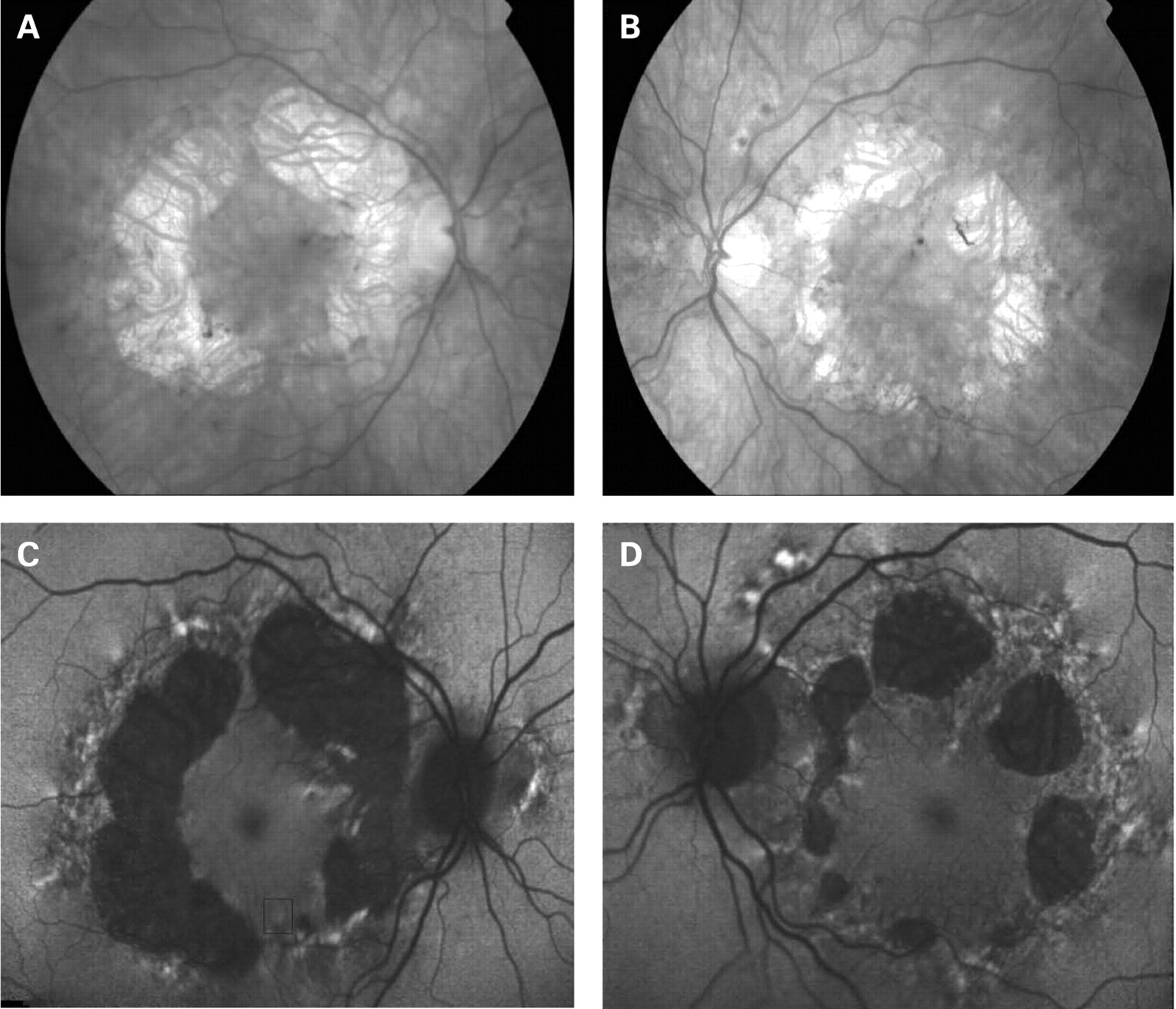
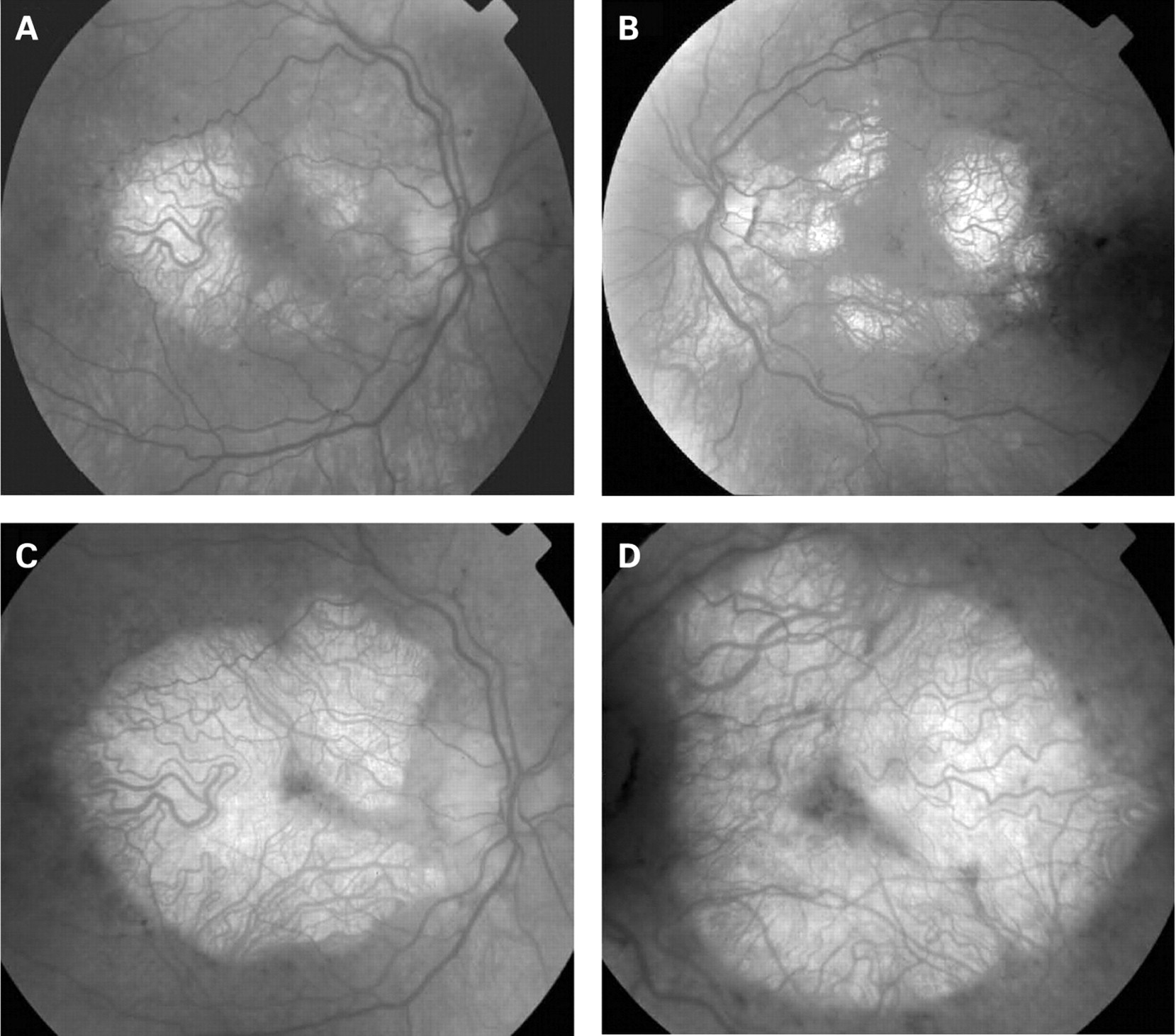
The fundus appearance was variable between patients, but two separate phenotypes were identified. The most common phenotype, occurring in nine of the 12 patients, was discontinuous perifoveal atrophy that was circumferentially distributed and oriented (fig 1). In patients who had follow-up over many years, the atrophy coalesced into a ring (fig 2). The central fovea was spared in all but one eye of a single patient (fig 3). Adjacent to the areas of atrophy were pale deposits at the level of the retinal pigment epithelium (RPE), granularity of the RPE and subretinal pigment clumping. The second phenotype, present in three patients, was an appearance consistent with a pattern dystrophy. In these three patients, no significant atrophy was present in the perifoveal area. There was granularity of the RPE and pale deposits and pigment clumping at the level of the RPE (fig 4). The majority of the fundus changes occurred within the temporal vascular arcades; however, RPE changes were also seen outside the arcades and nasal to the optic nerve.




The appearance on fundus autofluorescence (AF) imaging was of a diffuse macular abnormality. Decreased AF was present in areas of atrophy, and the pale deposits revealed increased AF. In the more common perifoveal atrophy phenotype, the retina surrounding the atrophic areas demonstrated speckled AF (figs 1, 3, 5). In the pattern dystrophy phenotype, there was a diffuse speckled appearance of the macular AF (figs 4 and 6). In neither case was the diffuse nature of the abnormality evident on biomicroscopy.
DISCUSSION
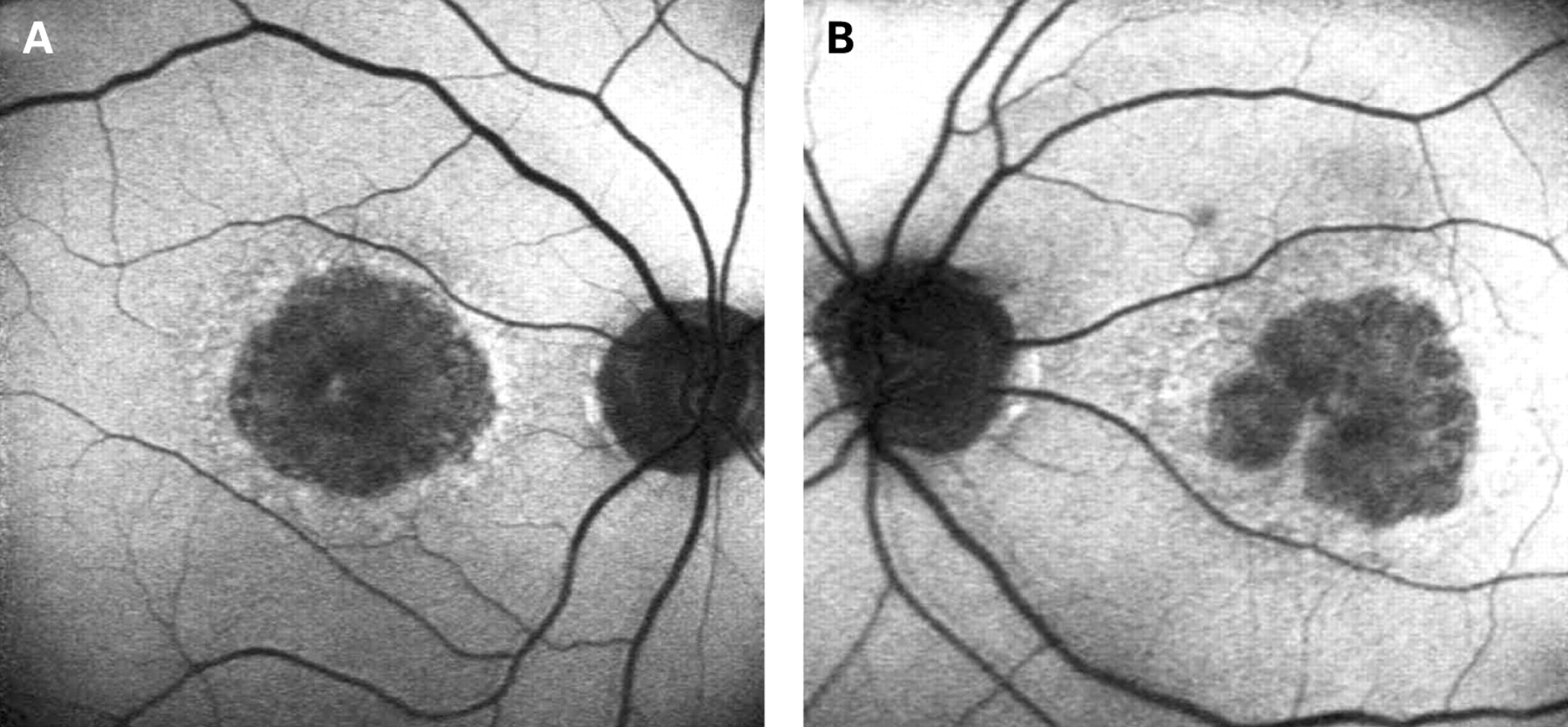
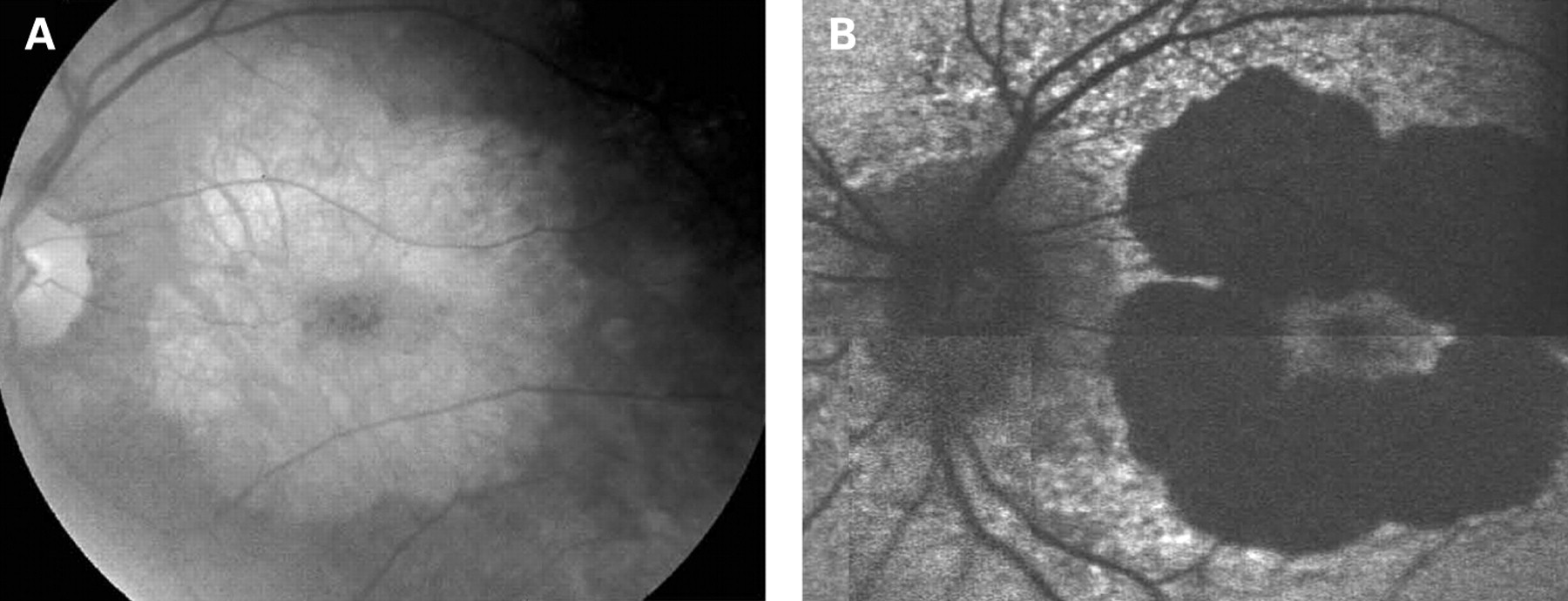
The characteristics of AF associated with the A3243G mtDNA mutation are distinct and differ from the majority of other macular dystrophies. In Stargardt macular dystrophy (STGD), well-defined atrophy is associated with diminished AF and the flecks with increased AF. The intervening retina has homogeneous AF. Figure 7A,B shows AF imaging of a male with typical fundus findings of STGD. In pattern dystrophy, as with STGD, the areas of abnormal AF are limited to abnormal areas detectable by ophthalmoscopy. In geographic atrophy (GA) due to age-related macular degeneration (AMD), the atrophic area is associated with decreased AF and may have a rim of increased AF as described by Holz et al (fig 8).20 In each of these conditions, the AF abnormalities correlate with the funduscopic abnormalities, and there is no widespread speckled AF, as observed with the A3243G mtDNA mutation. In the latter, it is not only the area of clinically detectable disturbance that is abnormal; indeed the area of abnormal AF is significantly larger than would be expected from the funduscopic appearance.

Perhaps the most difficult macular dystrophy to distinguish from that associated with the A3243G mtDNA mutation, by AF imaging alone, is the maculopathy caused by the dominant R172W peripherin mutation. The AF imaging in patients with the R172W mutation has been previously described and appears to depend on the stage of the disease.21 In the early symptomatic stages, these patients have a diffuse macular abnormality on AF consisting of speckled areas of increased and decreased AF within the macula simulating the pattern dystrophy-like phenotype of the A3243G mtDNA mutation. Later areas of atrophy develop within the areas of abnormal autofluorescence although not in the circular pattern seen in the patients with the mitochondrial dystrophy in this series. In addition, unlike A3243G maculopathy, the changes seen in R172W patients appear to be confined to the macular and peripapillary regions until very late in the disease when atrophic changes can extend beyond the arcades (fig 9).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The estimated prevalence of the A3243G mtDNA mutation resulting in MIDD in the diabetic population varies between 0.13 and 2.8%.30–38 In a recent multi-centre study, the systemic manifestations of MIDD in patients with diabetes were recorded in order to ascertain patients with diabetes who would benefit from screening for mitochondrial mutations.29 All patients included in this study had diabetes, 98% had bilateral neurosensory hearing loss, and 87% had macular pattern dystrophy. The percentage of patients with diabetes and deafness in this group was very high, as would be expected, given that patients were enrolled in the study based on these traits. In general, patients with the A3243G mtDNA mutation have a variety of disease manifestations including MIDD.5 6 39–41 Patients with macular dystrophy associated with the A3243G mutation diagnosed from an ophthalmology clinic may have more variable systemic manifestations than patients ascertained based on the presence of diabetes and deafness. In our small cohort of patients who presented with macular dystrophy, 83% (10/12) had symptomatic hearing loss, while only 33% (4/12) had diabetes. None of the patients in this series had diabetes without hearing loss. In addition, all patients in this series were diagnosed by the ophthalmologist as having a mitochondrial macular dystrophy, which was confirmed by testing for the A3243G mutation.
A potential weakness of this paper is the fact that only patients positive for the mitochondrial DNA mutation were included in the study. Although some patients were probably tested for the mutation and found to be negative, those patients were not included in this analysis. Therefore, we do not know the number of patients seen during the study period with a similar clinical phenotype who were negative for the A3243G mutation. Additionally, we assume the AF pattern we describe would be the same in all patients with a macular dystrophy secondary to the A3243G mutation. But because all of our patients were seen in an ophthalmology clinic, it might be interesting to perform a similar study on a population of diabetics known to have the A3243G mitochondrial DNA mutation to ascertain any differences in AF patterns in these patients.
The actual incidence of macular dystrophy associated with the A3243G mutation is probably not known, as most studies, including ours, have some selection bias based on the clinical setting from which the patients are ascertained. But certainly the incidence of macular dystrophy is probably much less common than the overall incidence of the A3243G mutation especially given the wide variety of clinical manifestations associated with this mutation.
Of interest for future study may be genotyping patients known to have the A3243G point mutation with and without evidence of macular dystrophy to determine if there is an increased incidence of genes known to increase AMD risk, such as complement factor H polymorphisms, in those patients with macular dystrophy compared with those without.
We conclude that the macular dystrophy associated with the A3243G mtDNA mutation has a recognizable phenotype by fundus autofluorescence in most cases and should be considered in the differential diagnosis, even in the absence of a personal or family history of diabetes and hearing loss.
Acknowledgments
We are grateful to the patients who kindly agreed to take part in this study.
REFERENCES
Footnotes
PR and SJ contributed equally to this study.
Funding: Financial support was from the Foundation Fighting Blindness and the Moorfields Special Trustees.
Competing interests: None.
Ethics approval: The study was approved by the local Ethics Committee.