Article Text

Abstract
Aims To describe subretinal debris found on ultrastructural examination in an eye with macular telangiectasia (MacTel) type 2 and on optical coherence tomography (OCT) in a subset of patients with MacTel type 2.
Methods Blocks from the mid-periphery and temporal perifovea of an eye with clinically documented MacTel type 2 were examined with electron microscopy (EM). Cases came from the Sydney centre of the MacTel project and the practices of the authors.
Results On EM examination, subretinal debris was found in the perifovea with accumulation of degenerate photoreceptor elements in the subretinal space. Despite the substantial subretinal debris, there was minimal retinal pigment epithelial (RPE) reaction. Focal defects were seen in the inner limiting membrane in the perifovea. Of the 65 Sydney MacTel project participants, three (5%) had prominent yellow material at the fovea. OCT revealed smooth mounds between the RPE and the ellipsoid region. The material was hyperautofluorescent.
Conclusions This study suggests that subretinal accumulation of photoreceptor debris may be a feature of MacTel type 2. Ultrastructural and OCT evidence of disease beyond the vasculature, involving photoreceptors and Muller cells, is presented.
- Retina
- Macula
- Pathology
- Imaging
- Degeneration
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Macular telangiectasia (MacTel) type 2, also known as idiopathic perifoveal telangiectasia, is characterised by loss of retinal transparency in the temporal juxtafoveal region and telangiectatic vessels with mild, diffuse leak on fluorescein angiography (FA) in its early, or non-proliferative phase.1 Other characteristic features include superficial crystals, the presence of foveal yellow deposits and focal juxtafoveolar pigment changes.2 Later clinical features include inner and outer lamellar cavitation with preservation of the inner limiting membrane (ILM)3 and, in some cases, retinal pigment epithelial (RPE) depigmentation, RPE atrophy and subretinal neovascularisation.4 The prevalence of MacTel type 2 was reported to be approximately 1 : 1000 in the Beaver Dam population older than 45 years,4 although this is likely to be an underestimate because the diagnosis was made from colour photographs that show only the more advanced features of the disease. MacTel type 2 is currently untreatable unless subretinal neovascularisation occurs. Its aetiology remains obscure, thus limiting strategies for prevention and treatment of the visual impairment it often causes.
Ultrastructural findings in MacTel type 2 are limited to one case.5 In this report we present novel ultrastructural observations in an eye with MacTel type 2 documented during life. One of these unexpected findings, the accumulation of subretinal debris, was also found on review of clinical imaging in a subset of patients with MacTel type 2.
Methods
Electron microscopy
The left eye of a 65-year-old man, diagnosed with MacTel type 2 in October 1987, was enucleated 4 h after death due to stroke in March 1999. Detailed history and clinical findings are described elsewhere.
Macroscopic examination before fixation in half-strength Karnovsky's fixative showed no other retinal abnormalities. Blocks from the mid-periphery and temporal perifovea were fixed and processed for electron microscopy (EM). Ultra thin sections were cut at 120 nm and stained with uranyl acetate 2% and Reynold's lead citrate. EM was carried out using a FEI Morgagni 268D transmission electron microscope (Eindhoven, The Netherlands) at 80 kV and digital images were acquired by an integrated MegaView III CCD camera (Olympus Soft Imaging Solutions, Munster, Germany).
Patients
Patients with subretinal debris were identified by review of spectral domain optical coherence tomography (OCT) images from the Sydney centre of the MacTel project and from the private practices of the authors.
Clinical examination and imaging
All patients underwent a complete ophthalmic examination including best corrected visual acuity testing, slit lamp examination with indirect ophthalmoscopy and OCT (Carl Zeiss Meditec, built-in software V.5.0). Cirrus HD OCT captures Macula Cube Scan 512 A-scans and 128 horizontal lines B-scans. A cross hair scan is taken automatically as part of the Cube Scan (1024 A-scan resolution).
Fundus autofluorescence imaging was performed using a confocal scanning laser ophthalmoscope (cSLO, HRA2; Heidelberg Engineering, Heidelberg, Germany, software V.1.5.90.2006). Fundus photography and FA was performed using the Topcon TRC-50 series at 35° with IMAGEnet 2000.
Results
Electron microscopy
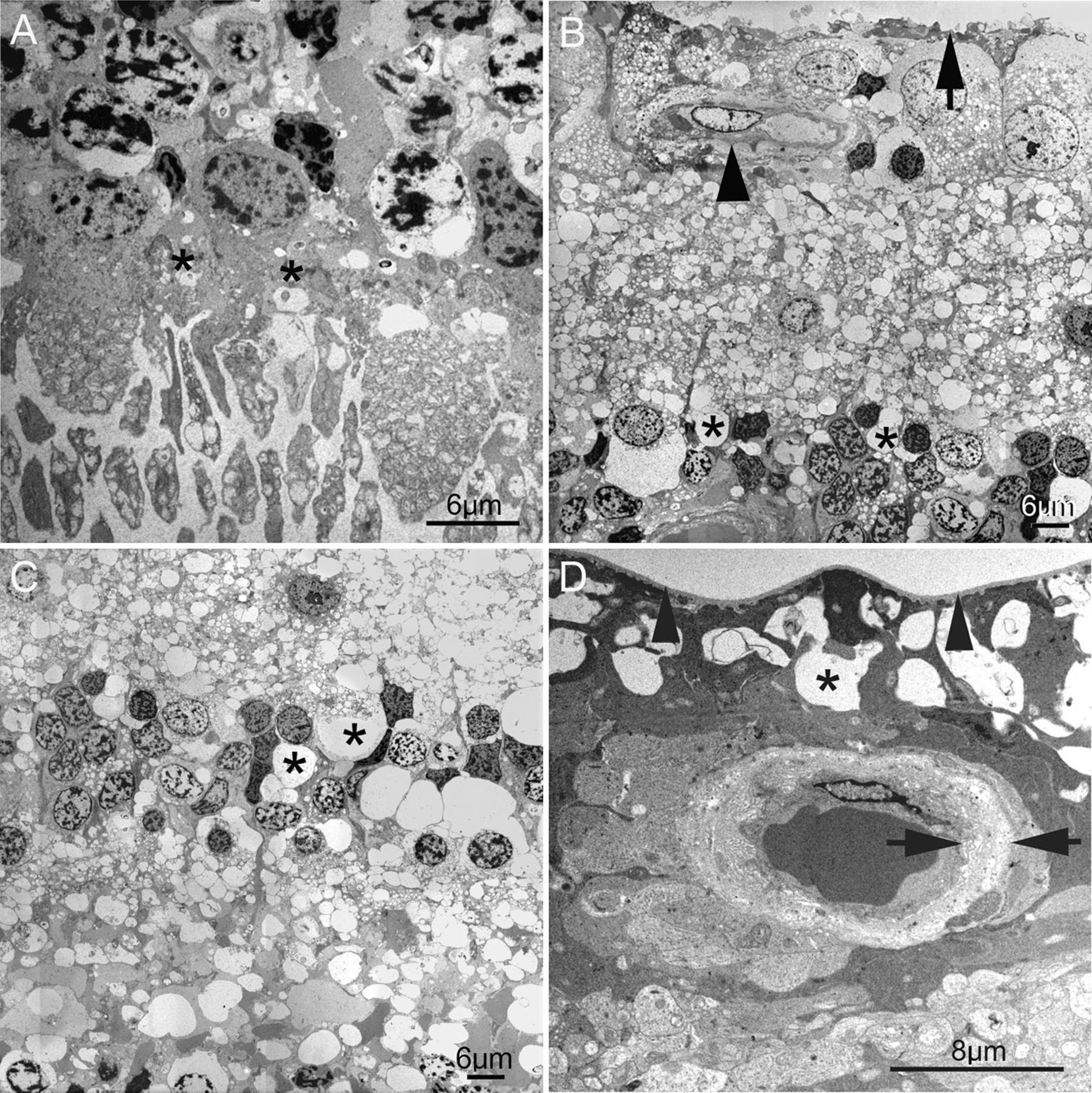
Vacuolar degeneration was seen in all layers of the retina, most notably in the outer plexiform layer in the perifoveal block (figures 1A,B). Empty spaces were noted in the inner nuclear layer, and dark, angulated Muller cell nuclei were sparse (figure 1A). Although these observations may be agonal in nature, the extent of changes in this eye was not present in other eyes with a 4 h postmortem delay from the same tissue bank.

(A) In this semithin section, ganglion cell density, a closely packed monolayer to the left and becoming bilayered to the right, show that the electron microscopy tissue block is from the outer perifovea. Vacuoles can be seen in all retinal layers, with empty spaces most noticeable within the inner nuclear layer. The subretinal debris is seen to the right of the image (arrows). With the exception of a single anteriorly migrated cell (arrowhead), the retinal pigment epithelium (RPE) remains a monolayer, despite the large amount of overlying debris. Small pigment epithelial detachments are seen on low power (centre and left). (B) Vacuolar change is present in outer nuclear photoreceptor cell bodies and nuclei, as well as in the outer plexiform layer in the perifovea (asterisks). At this magnification, the subretinal debris appears to consist of shed photoreceptor outer segments (arrows) in contact with the apical membranes of the RPE. The arrowhead identifies the subretinal space. (C) Higher magnification of the subretinal debris shows little RPE reaction, with a single anteriorly migrated RPE cell. There is minimal RPE hypertrophy or hyperpigmentation. (D) The RPE plasma membrane (black arrowhead) is separated from its basement membrane (white arrowhead) by empty space, probably originally containing fluid, giving rise to the pigment epithelial detachments seen on low power in (A). In a minority of instances, the RPE is separated from its basement membrane by irregular basal foldings of its plasma membrane (inset). Bruch's membrane in the perifovea appears unremarkable. (D) Intracytoplasmic vacuoles suggestive of autophagy within RPE from the mid-periphery. Again, no abnormalities are seen within Bruch's membrane in the mid-periphery.
Low magnification images showed substantial subretinal debris with minimal RPE disturbance and small pigment epithelial detachments (figure 1A). On higher magnification, the subretinal debris appeared to consist of degenerate photoreceptor outer segments (figure 1B), forming a thick layer over the apical surface of the RPE (figures 1C,D). Despite this, there was only mild RPE hyperpigmentation with very focal anterior migration (figure 1A,C,D). Two other previously unreported RPE changes were observed ultrastructurally. In the perifovea, content-poor sub-RPE deposits with sloping sides were seen, forming small pigment epithelial detachments resembling soft drusen (figure 1D). However, their location—between the RPE plasma membrane and its basement membrane—differed. A minority of these sub-RPE spaces were found on higher magnification to consist of basal infoldings of RPE plasma membrane (figure 1D inset). In the mid-periphery, large intracytoplasmic spaces were seen within RPE, possibly representing autophagic vacuoles (figure 1E).
Bruch's membrane and the choroid in the mid-periphery and perifovea showed no significant abnormalities (figure 1C–E).
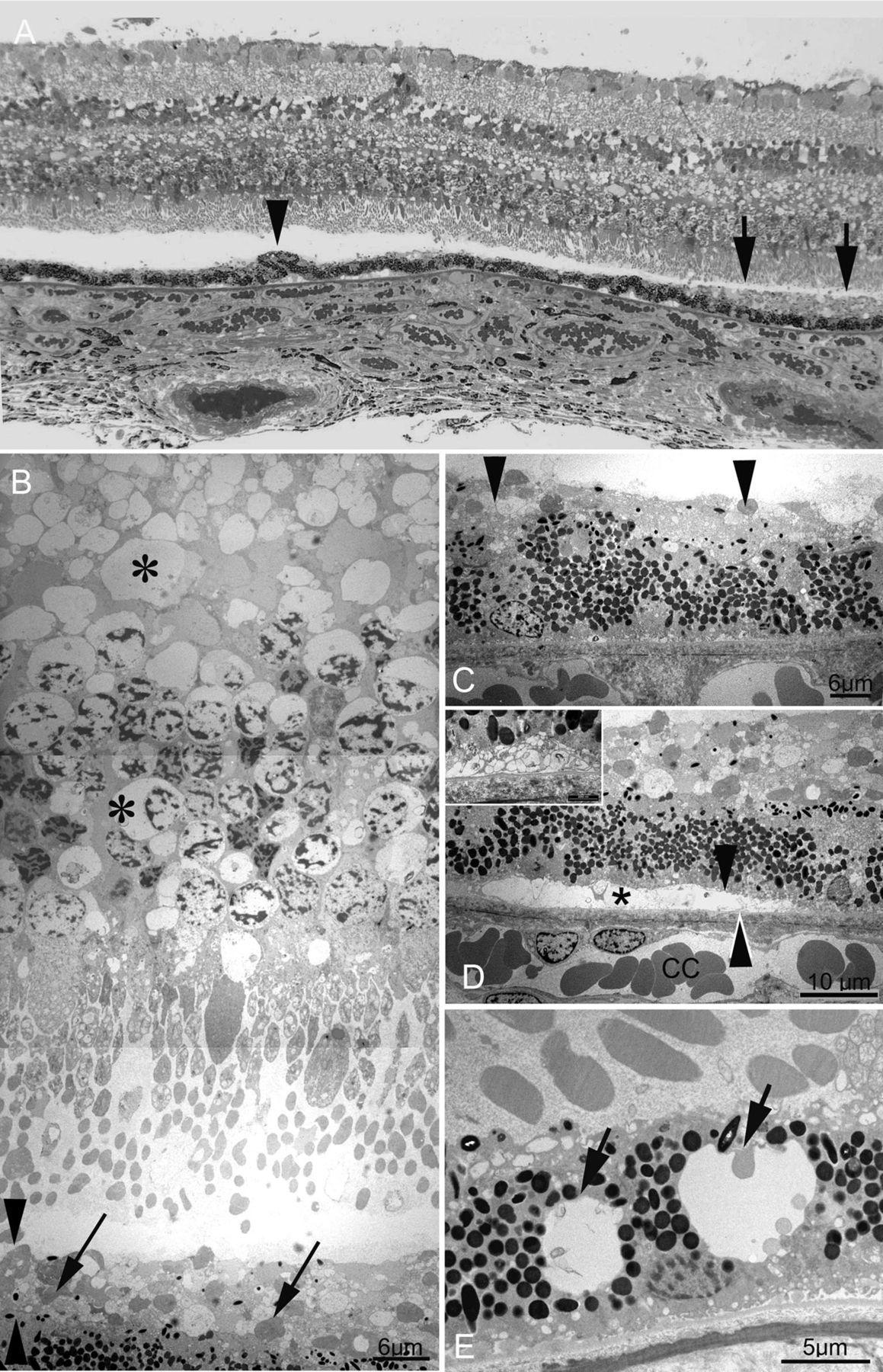
The outer limiting membrane (OLM) showed focal disruption, with absence of zonula adherens junctions between photoreceptors and Muller cell processes (figure 2A). There was no evidence of the replacement of lost photoreceptor–Muller zonula adherens junctions with Muller–Muller junctions, and no evidence of gliosis. Higher magnification of the inner nuclear layer showed empty spaces, possibly representing lost nuclei (figure 2C). The dark, intended nuclei of Muller cells are sparse in the inner nuclear layer (figure 2C). The ILM showed mild irregularity in the perifovea (figure 2B) but appeared intact in the mid-periphery (figure 2D).
(A) Focal loss of the outer limiting membrane (OLM) in the perifovea, evidenced by the absence of zonula adherens junctions between photoreceptor cell bodies and Muller cell processes (between asterisks). (B) In contrast to the OLM, the inner limiting membrane (ILM) within the perifovea appears largely intact (arrow). Prominent vacuoles are seen in the inner plexiform layer. A perifoveal retinal vessel shows subendothelial thickening and basement membrane reduplication (arrowhead). (C) Higher magnification of the inner nuclear layer from the perifoveal block shows empty spaces possibly representing lost nuclei (asterisks).The dark, indented nuclei of Muller cells appears sparse. (D) A retinal vessel from the mid-peripheral block, showing subendothelial thickening, with basement membrane reduplication (arrows). The ganglion cell layer and nerve fibre layer in the mid-periphery show empty spaces (asterisk), possibly representing lost ganglion cells or atrocytes. The ILM here is intact (arrowhead).
Previously described subendothelial thickening of retinal vessels were seen in both perifovea (figure 2B) and mid-periphery (figure 2D). These vascular changes are likely not to be specific for MacTel type 2, as vacuolation and lamination of the retinal capillary basement membrane may occur in otherwise normal eyes with age.6
Clinical cases
Patient characteristics
Patient characteristics are summarised in table 1. Of the 65 participants in the Sydney centre of the MacTel project Natural History Observation study that were reviewed systematically, three (5%) had prominent subretinal deposits that were visible on biomicroscopy. A further three cases were identified from the practices of the authors. The diagnosis of MacTel type 2 was confirmed for the first three cases by the Reading Centre at Moorfields Eye Hospital, London. The diagnosis in cases 4–6 was made on the basis of typical clinical findings in both eyes, including loss of macula transparency, vascular telangiectasis with late leak seen on FA, loss of luteal pigment seen with fundus autofluorescence (figure 3) and, in some cases, characteristic OCT features such as inner retinal cavitation.
Summary of the clinical features of the six cases

Photographic studies of case 1. Red free photographs of right (A) and left (B) eyes, arrow indicates debris beneath the right fovea. Early and late phase angiograms of the right (C and E) and left (D and F) eyes displaying typical features of MacTel type 2. Autofluorescence studies of right (G) and left (H) eyes. Arrows indicate hyperautofluorescent material beneath the fovea of both eyes, which was only evident clinically in the right eye.
The ophthalmic and medical histories of the six cases were generally unremarkable. Case 3 was mildly myopic. A history of hypertension was reported by three cases, skin malignancy by two, dyslipidaemia by two, type II diabetes by two and gout by one case. This profile of systemic disease is similar to that reported by the MacTel project Natural History Study.7
Imaging findings
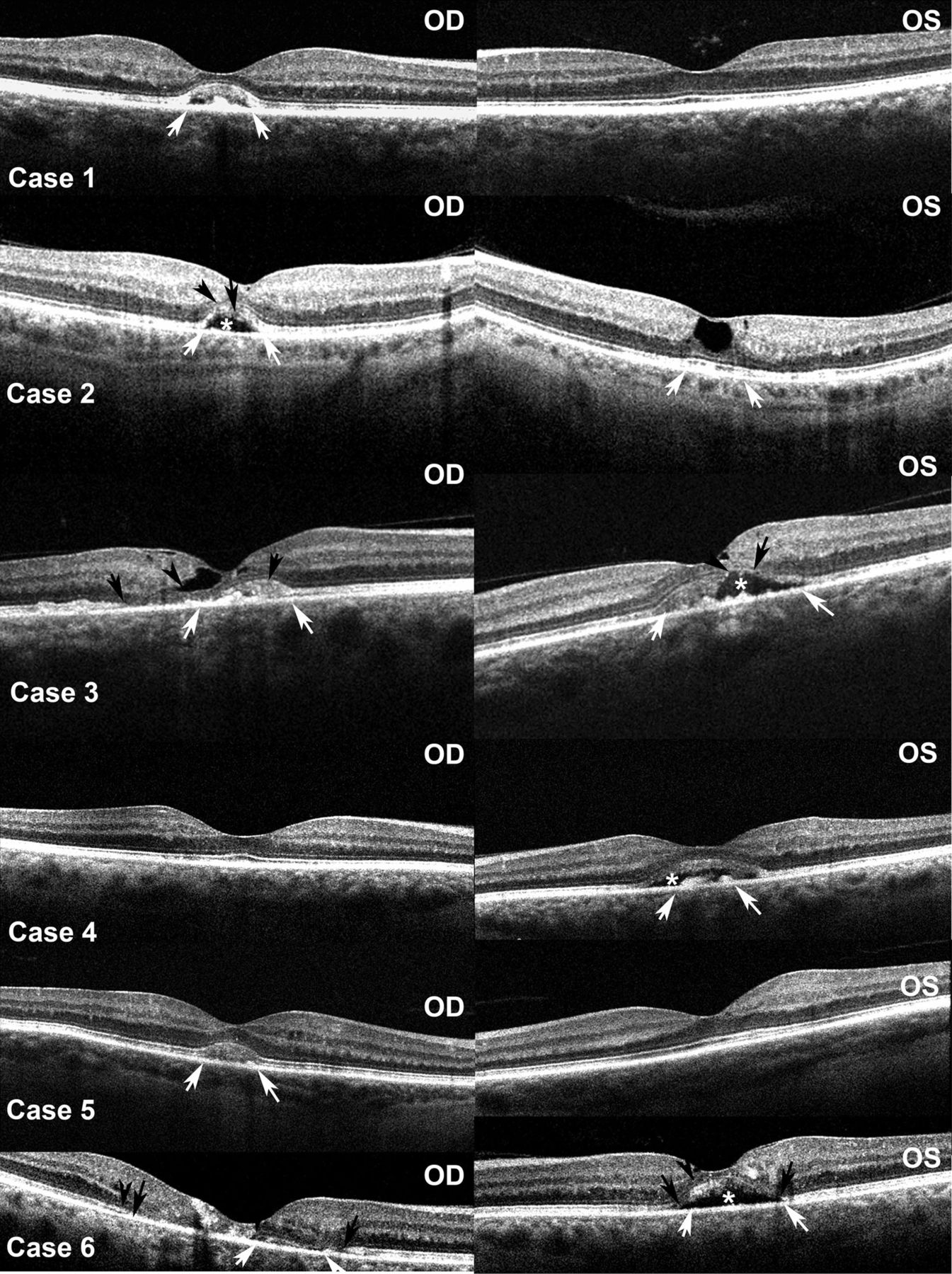
OCT scanning of all eyes with biomicroscopically visible subretinal debris revealed a prominent mound of material between the ellipsoid region and the RPE, with irregular areas of hyporeflectivity to a variable extent (figure 4). There was irregular reflectivity and focal loss of the photoreceptor layer over the surface of most of the deposits, with the OLM similarly affected to a somewhat lesser effect. The apex of the dome of the subretinal material was beneath the centre of the fovea in all affected eyes except the right eye of case 3 in which it was displaced slightly nasally.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Optical coherence tomography studies of both eyes of the six cases. Open arrows indicate margins of the subretinal debris where present; closed arrows indicate areas of photoreceptor disruption; arrowheads indicate discontinuities in the outer limiting membrane; asterisks indicate cavitation within the subretinal debris.
The subretinal material did not affect the MacTel type 2 phenotype. Patients were of the typical age, the visual acuity was mildly to modestly affected (table 1). Pigment clumping beneath the debris was detected in only one of eight affected eyes. The subretinal deposit was unilateral in four of the six cases. In all cases the subretinal material was hyperautofluorescent and did not have any distinguishing qualities on angiography. Neither hypo nor hyperfluorescence was seen in the early or late stages, although the latter were difficult to interpret due to the generalised diffuse late hyperfluorescence that ciIn cases 4 and 5. In the other six pairs of eyes it was similar.
Discussion
Gass8 noted that approximately 5% (precisely the incidence we report here) of people with MacTel type 2 had a yellow, round ‘intraretinal’ spot 100–300 microns in diameter at the centre of the foveola of one or both eyes that was associated with slight loss of the foveal depression, but without OCT he was unable to speculate on what caused it. With the benefit of a rare specimen and modern imaging techniques, it appears that the material, which is subretinal, probably represents degenerate photoreceptor outer segments.
The material we describe in the MacTel type 2 cases is similar to that found with foveomacular vitelliform dystrophy (FMVD) with some significant differences. One or both eyes may be affected in both conditions by similar-looking yellow material, which is external to the IS:OS break as shown by spectral domain OCT. Loss of luteal pigment in MacTel type 2 results in loss of masking of central autofluorescence, but it does not cause a stronger signal compared with the autofluorescence in the outer macula such as was seen in these cases. This hyperautofluorescence, which is also seen in FMVD, suggests a common origin from the outer segments of photoreceptors, which contain lipofuscin-associated pigments. However, FMVD is almost invariably associated with clumping of the RPE, which we found in only one of eight affected eyes. FMVD material is hypofluorescent in the early stages and stains late on FA; these features were not found in any of the MacTel type 2 cases we present, perhaps because the outer blood–retinal barrier remains intact. Ultrastructurally, the material found in both MacTel type 2 and FMVD is composed of photoreceptor outer segments. However, in FMVD, there is an obvious RPE reaction to the material, with RPE hypertrophy, hyperpigmentation and/or anterior migration.9 In the MacTel type 2 eye, this reaction was essentially absent.
Despite significant vacuolar change of the neural retina and focal loss of the OLM, there was no ultrastructural evidence of reactive gliosis. Muller cells are neuroprotective and also maintain the vascular and structural integrity of the neural retina. Muller cell gliosis occurs in response to both acute and chronic retinal injury and stress.10 ,11 This response is seen in all major retinal diseases, including diabetic retinopathy12 and age-related macular degeneration.13 ,14 An early event in Muller cell gliosis is expression of the intermediate filament glial acidic fibrillary protein (GFAP), which is thought to increase the structural integrity of the retina. Of note, immunofluorescence performed on the fellow eye of the same patient in an earlier study showed a reduction in macula Muller cells and a striking absence of Muller cell GFAP expression.6
Similarly, in the context of subretinal accumulation of photoreceptor debris, the minimal RPE reaction observed in the eye examined by EM and in all except one of the clinical cases is unexpected. An increased phagocytic load (and other stimuli such as trauma, inflammation or intraocular tumour) typically leads to hypertrophy, hyperpigmentation and anterior migration of the RPE. Although RPE–Muller cell interactions are poorly understood, there is evidence that the RPE response depends, to some extent, on normal Muller cell function. RPE changes in age-related macular degeneration, for example, have been shown to occur in the setting of Muller cell GFAP expression and activation.15 In the Royal College of Surgeons rat model of retinal degeneration, an RPE reaction to accumulated subretinal photoreceptor debris is only observed in areas of the retina where GFAP-expressing Muller cells are preserved.16 It is plausible, therefore, that the attenuated RPE reaction in MacTel type 2 eyes may also be the result of defective Muller cell function.
While obvious subretinal debris was only seen in 5% of MacTel study participants, we believe it is possible that subclinical debris may be present in many MacTel type 2 eyes where it may reflect an underlying process of impaired phagocytosis by the RPE. In the left eye of case 1 the material was only evident on autofluorescence imaging, not on fundoscopy. We are unable to say at what stage in the condition the subretinal material might become apparent. The clearance of photoreceptor debris in the outer retina depends primarily on the RPE and on Bruch's membrane permeability, with a probable contribution by retinal microglia.17 It is unlikely that reduced Bruch's membrane permeability is the main pathology in MacTel type 2 eyes, as there was no evidence of thickening or accumulation of lipid and membranous material within Bruch's membrane. The inability to clear the subretinal deposits in some MacTel type 2 eyes must therefore be due to: (1) phenotypic variation in the phagocytic potential of RPE or microglia; or (2) disrupted Muller cell–RPE and/or Muller cell–microglia interactions.
The fact that the vascular changes were symmetrical in the six cases in which the material was detected in only one eye suggest that the excessive accumulation of subretinal debris seen in the clinical cases is independent of, rather than caused by, the vasculopathy. This too would be consistent with an upstream Muller cell defect, which might be expected to affect blood vessels and photoreceptors independently.
The clinicopathological correlation in this report is limited by the lack of autofluorescence and OCT findings in the eye examined by EM and by the absence of tissue for histological or ultrastructural examination for the clinical cases. However, both clinical and ultrastructural observations demonstrate that the accumulation of subretinal debris is a feature in a subset of patients with MacTel type 2 and may further implicate Muller cells in the pathogenesis of the disease.
References
Footnotes
-
Contributors All listed authors have contributed substantially to study conception/design, data acquisition/analysis or interpretation, manuscript drafting/revision, and all have approved the final draft submitted for publication.
-
Funding This study received funding from the Lowy Medical Research Institute.
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.