Article Text

Abstract
Aim To evaluate efficacy and safety of intravitreal aflibercept injection (IAI) in subjects who were previously treated with ranibizumab and/or bevacizumab for active exudative age-related macular degeneration (AMD).
Methods Patients (n=26) were enrolled in a 12-month prospective, interventional, single arm, investigator-initiated study with planned 6-month interim analysis. Patients with active exudative AMD, previously treated with ranibizumab and/or bevacizumab, were treated with 2 mg IAI every month for the first 3 months, followed by a fixed dosing schedule of 2 mg IAI every 2 months. The primary study endpoint was the mean absolute change from baseline central subfield thickness (CST) at month 12 as measured by SDOCT. Secondary outcomes included mean change from baseline best-corrected visual acuity (BCVA) score, percentage of subjects who gained or lost greater than or equal to 15 letters of vision, percentage of subjects who are 20/40 or better, percentage of subjects who are 20/200 or worse, and the incidence of adverse events (AE) and serious AEs.
Results Planned 6-month interim analysis demonstrated a mean decrease in CST of 38.6 µm (p<0.001) and a mean increase in ETDRS BCVA of +5.9 letters (p<0.001). Fifteen percent of subjects experienced a greater than 15-letter improvement in visual acuity, 84.6% of patients gained visual acuity, and no patient lost 3 lines of vision from baseline. Forty-two percent of subjects were 20/40 or better, and 11.5% of subjects were 20/200 or worse at month 6. No serious ocular or systemic AEs were encountered.
Conclusions IAI-treated eyes demonstrated improved short-term functional and anatomic endpoints in subjects with active exudative AMD switching from previous anti-VEGF treatment when given in a fixed dosing scheme for 6 months.
Trial registration number NCT01617148.
- Clinical Trial
- Macula
- Neovascularisation
- Retina
- Angiogenesis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Age-related macular degeneration (AMD) is a leading cause of adult blindness in the developed world.1 Severe visual loss from AMD is caused by subfoveal geographic atrophy and choroidal neovascularisation, which is the hallmark of exudative AMD. Vascular endothelial growth factor (VEGF) is a growth factor that has been implicated as a major pathogenic factor in exudative AMD as it stimulates angiogenesis and increases vascular permeability.2 Inhibitors of VEGF have provided significant therapeutic benefit to subjects suffering from this disorder.3–5
Aflibercept is a recombinant fusion protein consisting of key human VEGF receptor extracellular domains from receptors 1 and 2 (VEGFR1 and VEGFR2) fused to the Fc domain of human IgG1.6 It is a dimeric glycoprotein with a molecular weight of 97 kDa that is ∼15% glycosylated, giving a total molecular weight of 115 kDa. Aflibercept has several theoretical advantages over other VEGF blockers: (1) it has a much higher binding affinity for VEGF (∼0.5 pM dissociation constant for VEGF165 and VEGF121) than either bevacizumab or ranibizumab7; (2) it binds related growth factors, such as placental growth factors 1 and 2 (PLGF1 and PLGF2) and VEGF-B, which may be advantageous in certain disease situations, including retinal neovascularisation8 and (3) the vitreous half-life of aflibercept (18 days) is longer than ranibizumab (9 days), but slightly shorter than bevacizumab (21 days).9
The pivotal, phase 3, VIEW studies that led to Food and Drug Administration approval required that enrolled subjects be naive of previous AMD treatments. Subjects were randomised to various intravitreal aflibercept injection (IAI) doses and dosing regimens, and compared to monthly ranibizumab treatment in a non-inferiority statistical design. Results revealed that subjects exhibited similar visual outcomes when comparing IAI 2 mg, dosed every 2 months after a 3-monthly loading dose, to monthly ranibizumab 0.5 mg at the primary outcome of 52 weeks.10 ,11 Thus, using IAI in this fashion reduced the number of injections without compromising visual outcomes compared to the gold standard of monthly ranibizumab.10
However, the majority of retina specialists do not treat using a fixed dosing pattern as used in the VIEW studies. Subjects are generally treated with as-needed dosing (pro ne rata, PRN) or on a treat-and-extend (TAE) paradigm. Studies, such as the Comparison of Age Related Macular Degeneration Treatment Trials (CATT) indicate that these treatment regimens are similar but not equivalent to fixed dosing.12 ,13 Moreover, the results of the HORIZON AMD, SECURE and SEVEN-UP studies demonstrate declines in visual acuity in subjects transitioned to PRN treatment from a fixed dosing scheme.14–16 The question still remains as to whether subjects would maintain visual gains if kept on a fixed dosing scheme.
Additionally, with the approval of any new anti-VEGF therapy, there are many subjects who have received previous anti-VEGF treatments and were switched to the new medication. There have been several case series describing the outcomes of switching from ranibizumab and/or bevacizumab to IAI with visual gains.17–20 The majority of these studies evaluated a recalcitrant or treatment-failure population. The purpose of this study is to prospectively evaluate patients with active exudative AMD responsive to anti-VEGF therapy who were switched to IAI using a similar fixed dosing scheme as used in the first year of the VIEW studies.
Materials and methods
Study design
The a single-arm, investigator-initiated study of the efficacy, safety and tolerability of intravitreal aflibercept injection in subjects (ASSESS) study is a prospective, interventional, single arm, investigator initiated study performed at the Cole Eye Institute, Cleveland, Ohio, USA. The study received approval from the Cleveland Clinic Investigational Review Board (IRB), and all study-related procedures were performed in accordance with good clinical practice (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) E6), applicable Food and Drug Association (FDA) regulations, and the Health Insurance Portability and Accountability Act. All subjects signed an informed consent for their participation in the study.
Participants
The main inclusion criteria were: (1) active subfoveal choroidal neovascularisation secondary to exudative AMD confirmed by fluorescein angiography; (2) Electronic - Early Treatment In Diabetic Retinopathy Study (E-ETDRS) vision of 25–80 letters (Snellen equivalent of ∼20/25–20/320); (3) at least one prior injection of 1.25 mg bevacizumab, or 0.5 mg ranibizumab (Avastin and Lucentis, respectively; Genentech, South San Francisco, California, USA) within 3 months of enrolment and (4) had an initial response on optical coherence tomography (OCT) defined as a decrease of retinal oedema and/or subretinal fluid to anti-VEGF injections followed by recurrent increase in fluid on OCT (further defined as intraretinal, cystoid, subretinal fluid, or worsening pigment epithelial detachment (PED)) or the presence of new haemorrhage on clinical examination. In general, the inclusion criteria intended to define a responder population to previous anti-VEGF treatment. This is in contrast with previous studies evaluating IAI that focused on a non-responding population.
The key exclusion criteria included any prior concomitant therapy with another investigational agent to treat neovascular AMD in the study eye, history of vitrectomy, trabulectomy, surgery for retinal detachment, or any intraocular or periocular surgery in the study eye (within 3 months of day 1), prior treatment with verteporfin (Visudyne, Valeant, New York, New York, USA) photodynamic therapy in the study eye, previous investigational treatments for AMD, history of subfoveal laser photocoagulation, uncontrolled glaucoma, or uveitis. Prior systemic anti-VEGF therapy was only allowed up to 3 months prior to enrolment, and was not allowed during the study. If the patient presented a history of vascular diseases affecting the retina within the study eye, they were excluded from the study.
Only one eye per subject was enrolled in the study. For subjects who met eligibility criteria in both eyes, the investigator and patient selected the study eye. If a subject's fellow (non-study) eye required treatment for exudative AMD at study entry, or during the subject's participation in the study, the fellow eye could receive IAI for wet AMD, but it was not considered as an additional study eye. The frequency of fellow eye treatment was based on investigator discretion.
Visits and assessments
Subjects were given 2 mg (0.05 mL) of IAI administered monthly for the first 3 months, followed by 2 mg (0.05 mL) once every 2 months as per the drug label (figure 1). Intravitreal aflibercept was supplied by Regeneron Pharmaceuticals, (Tarrytown, New York, USA) and was administered using the standard aseptic intravitreal techniques detailed in the package insert.21

ASSESS study design.
After enrolment, patients were seen and treated according to the study schema in figure 1. Evaluations occurred once a month for the first 3 months and every 2 months thereafter until month 12. At each visit, the visual functions of both the eyes were assessed using the E-ETDRS chart (M&S Systems) and protocol visual acuity measurement consisting of best corrected visual acuity testing and a forced choice paradigm.22 A comprehensive eye examination and spectral domain optical coherence tomography (SD-OCT) scanning were performed on both the eyes. The scanning protocol consisted of a macular cube scan as well as a series of high-definition 6.0 mm linear scans centred on the fovea using the Cirrus SDOCT (Humphrey Zeiss, San Leandro, California, USA, software V.6.0). Two independent graders scored the qualitative morphological patterns of the OCT scans. Central subfield thickness (CST) was measured using the automated software. Manual registration of pre-IAI, IAI and post-IAI OCT images was done in cases of inaccurate automated registration before obtaining the CST.
Study end points
The primary study endpoint was the mean absolute change from baseline in CST (defined as the average thickness within the central 1 mm subfield) at the month 6 planned interim analysis. The secondary outcomes included the mean change from baseline in best-corrected visual acuity (BCVA) score, change from baseline in macular volume and cube average thickness by SDOCT, the percentage of subjects who gained greater than 15 letters of vision, the percentage of subjects who lost greater than 15 letters of vision, the percentage of subjects who were 20/40 or better, and the percentage of subjects who were 20/200 or worse. The incidence of ocular and non-ocular adverse events (AE) and serious AEs were also documented.
Safety analyses
Safety was assessed through collection and summary of ocular and non-ocular AEs, systemic adverse events (SAE), and ocular assessments. The occurrence of any severe postoperative complications, including infection, inflammation, arteriothrombolic events, or death after the first IAI were recorded. At each study visit, non-directive questioning was performed by the study coordinator to elicit AE reports from subjects. All AEs and SAEs, whether volunteered by the subject, discovered by study site personnel during questioning, or detected by examination, laboratory testing, or other means, were recorded in the case report forms.
Statistical methods
Measures were summarised using means, SDs, median and range. Normality of measures was evaluated using the Shapiro–Wilk test. Since data were mostly normally distributed, comparisons with and between groups were performed using two-sided paired t tests. Where appropriate, sensitivity analyses using non-parametric Wilcoxon signed rank tests were also performed. Since groups significantly differed at baseline on both measures, a mixed-effect model was also fit, with change as the response, and eye (treated or control) as the primary predictor and baseline measure as a covariate. Analyses were performed using SAS software (V.9.2; Cary, North Carolina, USA). A significance level of 0.05 was assumed for all tests.
Results
A total of 26 subjects were included in the study (table 1). The average age of the subject was 78 years (range 69–90 years). The average length of time since diagnosis and first treatment of exudative AMD was 14 months±11 months, ranging from 3 to 35 months. Twenty-seven percent (7 subjects) were previously treated with bevacizumab, 65.4% (17 subjects) were previously treated with ranibizumab, and 7.1% of subjects were treated with both drugs. Patients were predominantly treated monthly PRN prior to study entry. The average number of anti-VEGF treatments prior to study entry was 9.6 injections (range 3–23 injections). Oftentimes, patients were referred by outside physicians, without complete documentation of how many previous anti-VEGF injections were given to the patient prior to study entry. This accounts for some patients who are documented to only have three previous anti-VEGF injections. Prior to study entry, average CST was 316 µm and average visual acuity was 20/63. The mean baseline E-ETDRS BCVA letter score upon entry was 56.4 letters (Snellen equivalent ∼20/80). The mean CST upon study entry was 304.1 µm (range 210–505 µm). Thirty-five per cent of subjects were 20/40 or better, and 23.1% of subjects were 20/200 or worse at study entry.
Baseline characteristics of patients enrolled within the a single arm, investigator-initiated study of the efficacy, safety and tolerability of intravitreal aflibercept injection in subjects trial
Figures 2A,B and 3A,B show summaries of anatomical and visual changes within the treatment group from baseline to 6 months. The mean BCVA score improved from 56.4 at baseline to 62.3 at month 6 (+5.9 letters). There was a significant improvement in BCVA at all follow-up visits except month 1 (M1: p=0.12, M2: p<0.001; M4: p=0.001; M6: p<0.001).
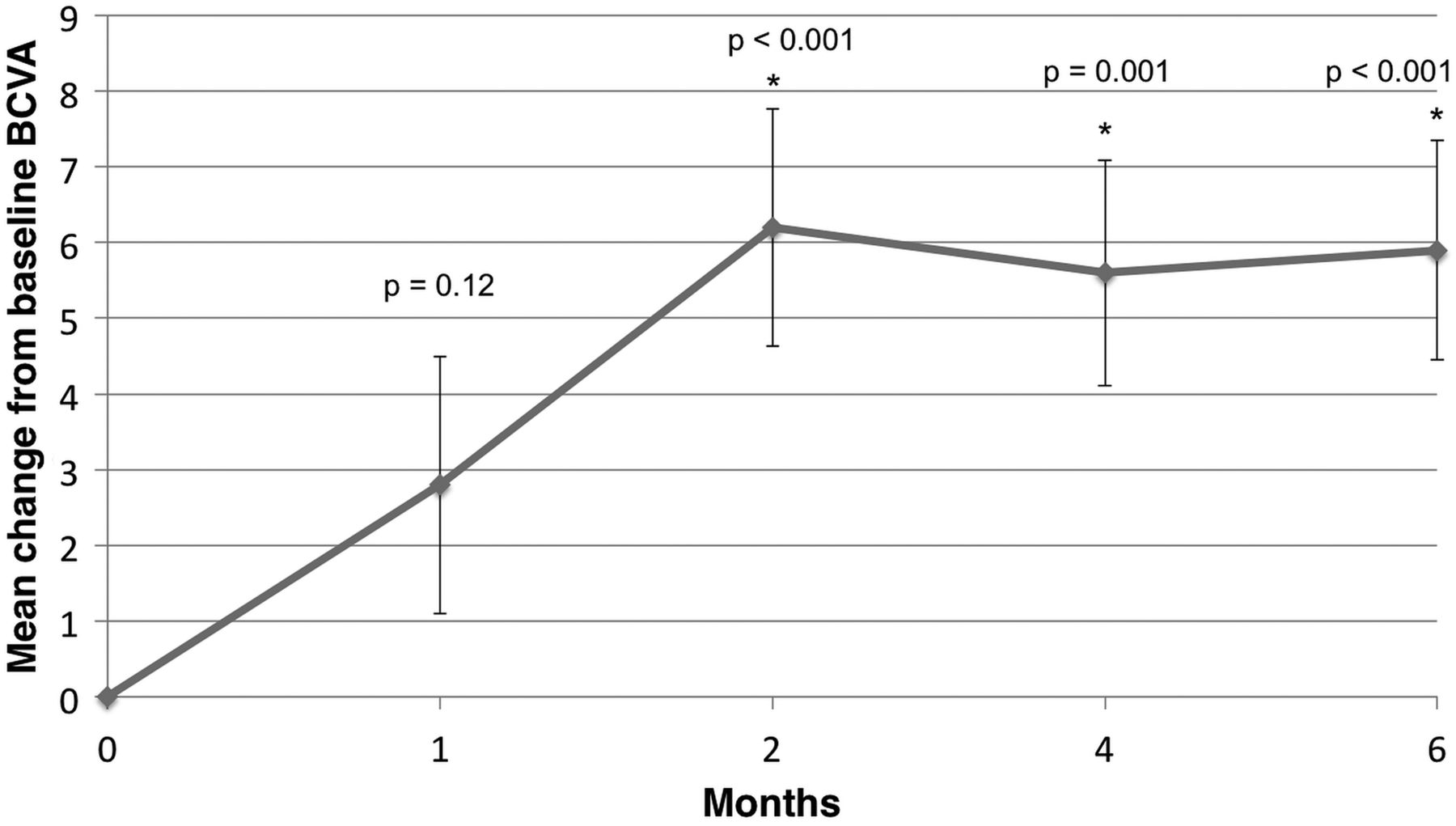
Mean change from baseline in best-corrected visual acuity (BCVA) score.

Mean change from baseline in central subfield thickness.
The mean CST improved from 304.1 µm to 265.5 µm at month 6 (−38.6 µm). CST was significantly improved from baseline in the study eye at all visits (p<0.001, p=0.001, p=0.003, and p<0.001, months 1, 2, 4 and 6, respectively). The average macular volume at month 6 was 9.8 mm3 (−0.5 mm3 reduction from baseline). There were statistically significant changes from baseline at all follow-up time periods (M1: p<0.001; M2: p<0.001; M4: p<0.001; M6: p=0.007). The mean cube average thickness at month 6 was 275.4 µm (10.8 µm reduction from baseline). All visits demonstrated significant improvements in cube average thickness in the treated eyes (M1: p<0.001; M2: p<0.001; M4: p=0.002; M6: p=0.021).
Figure 4 demonstrates the visual acuity changes in patients noted by month 6. Study results showed that 15.38% of subjects gained at least 15 letters of vision by month 6. There were no subjects who lost more than 15 letters of vision. One-hundred percent of subjects were considered visually stable (defined as the number of subjects who gained, were stable, or lost up to two lines of acuity). Thirty-nine percent of subjects were 20/40 or better at month 6, and 11.5% of patients were 20/200 or worse at month 6, an improvement from baseline in both categories.

Best-corrected visual acuity distribution at month 6.
In order to determine the changes in retinal anatomy, qualitative OCT grading of registered scans was performed to observe for the presence of subretinal fluid, intraretinal fluid (or cysts), and PED. A PED was defined as a highly reflective elevated band of retinal pigment epithelium overlying a less reflective cavity, which is almost optically empty. Two independent OCT graders performed all morphologic analysis and defined the changes from baselines as improved, stable, or worse for each OCT characteristic. Any disagreement between graders was given to a third grader for consensus determination.
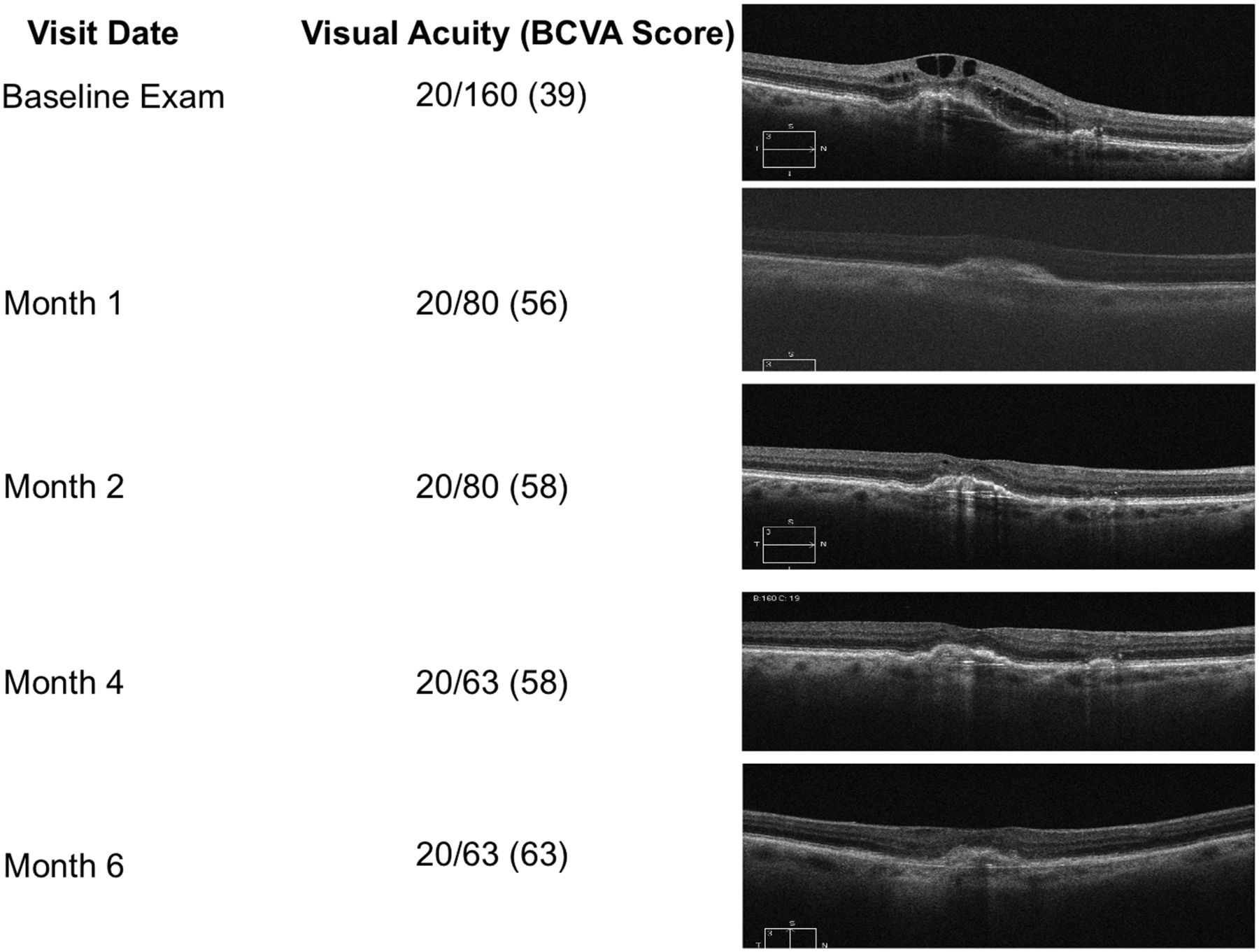
At the month 6 visit, 50% of eyes had an anatomical improvement in subretinal fluid; 34.6% were anatomically stable in subretinal fluid with comparison to their baseline visit, and 15.4% presented anatomically worsened subretinal fluid in comparison with their baseline visit (figure 5). At the month 6 visit, 34.6% of subjects presented anatomically improved intraretinal fluid in comparison with their baseline visit, 53.9% presented anatomically stable intraretinal fluid, and 11.5% presented anatomically worsened intraretinal fluid. With regards to PEDs, 19.2% of subjects presented anatomically improved PEDs in comparison with their baseline visit, 76.9% presented anatomically stable PEDs, and 3.9% presented anatomically worsened PEDs. Figure 6 is a case representation of a patient within the trial.
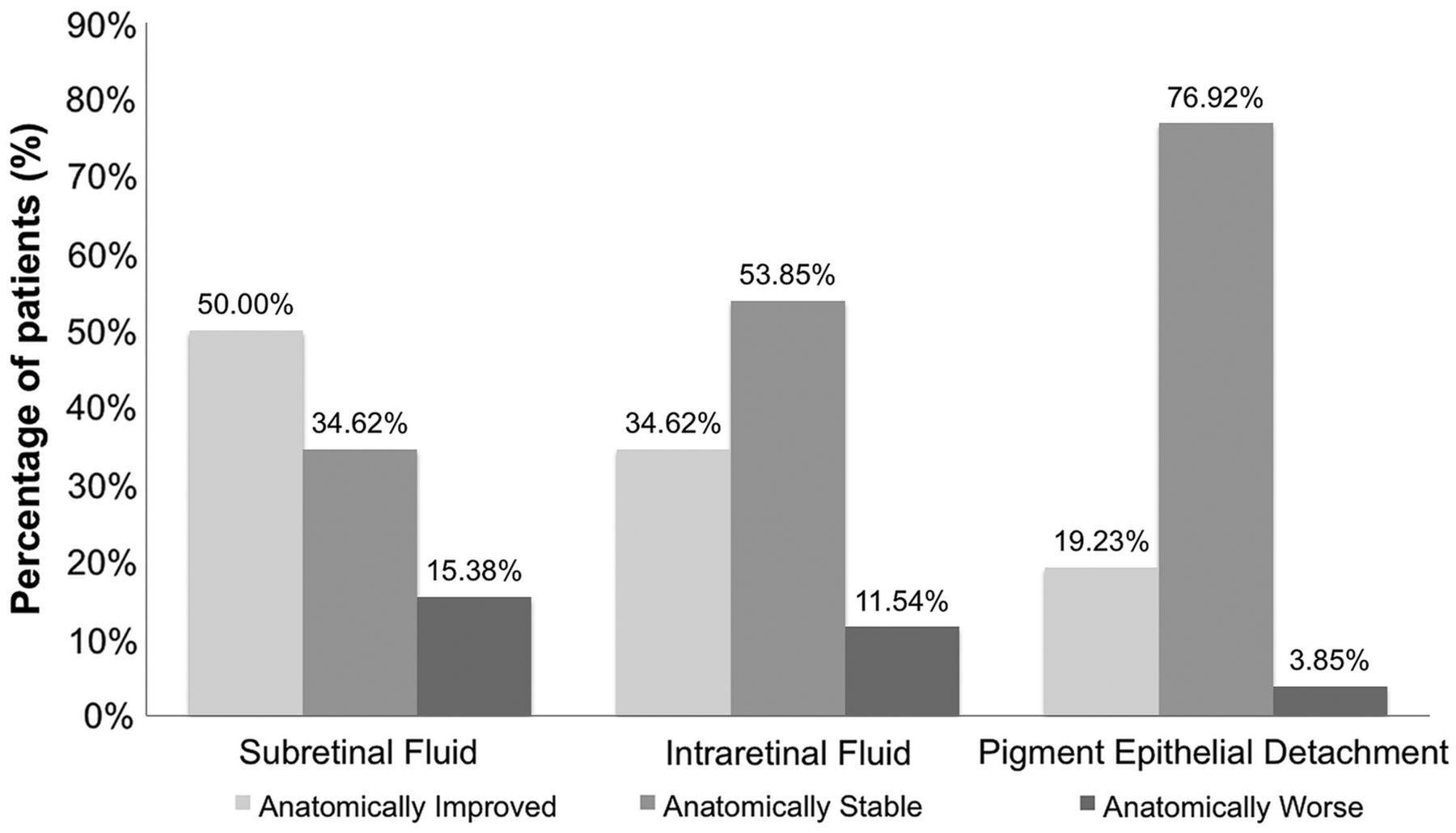
Optical coherence tomography fluid status in patients at month 6.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Representative patient from the ASSESS trial demonstrating treatment response.
There were no serious AEs during the first 6 months of the study.
Discussion
This study is the first prospective report examining the effect on subjects transitioned from other anti-VEGF agents to IAI for exudative AMD using a fixed dosing paradigm as per the FDA label. There was a statistically significant improvement from baseline measurements in BCVA, CST, cube average thickness and macular volume. This 6-month interim analysis demonstrated a mean decrease in CST of 38.6 µm (p<0.001) in the 26 previously treated eyes, and a mean increase in E-ETDRS visual acuity of +5.9 letters (p<0.001). All subjects were considered visually stable within the study, and the majority of subjects had either stable or improved anatomical OCT findings with regards to subretinal fluid, intraretinal fluid and PEDs.
It is important to realise that these subjects had, on average, received 9 previous anti-VEGF injections with an improvement or decline in visual acuity and OCT from when they were first diagnosed with wet AMD and started on anti-VEGF therapy. In the MARINA, ANCHOR, CATT, IVAN studies, very few subjects demonstrated improved vision after nine previous injections, irrespective of whether they received monthly or PRN treatment. In fact, in the CATT study, subjects’ vision declined when switching from a monthly to PRN regimen after 1 year.12 ,13 Thus, if the subjects continued with their previous anti-VEGF regimen, it is unlikely that the visual and anatomic improvement seen within this study would have occurred.
During the first 6 months of this study, there were no serious ocular or SAEs reported. The results of this 6-month study suggest that IAI is an effective treatment for AMD even after adequate response to previous treatment with ranibizumab and/or bevacizumab.
The current aim in the treatment of wet AMD is to maintain a ‘dry’ retina on OCT. However, recent studies have demonstrated that a small amount of fluid may not make a significant difference in visual outcomes.23 For example, in the CATT study, subjects in the PRN groups had more persistent retinal fluid on OCT (72.5% with monthly ranibizumab and 82.7% with monthly bevacizumab; p<0.001) despite having similar visual results. This finding was also seen in the HARBOR Trial.5 The present study confirmed some of these findings with the majority of patients manifesting a qualitative improvement but still incomplete resolution of intraretinal fluid, subretinal fluid and/or PEDs at 6 months, despite having a statistically significant gain in visual acuity.
The 6-month interim analysis of the ASSESS study supports the conversion of patients from PRN or TAE regimens to a fixed dosing regimen with IAI as per the FDA label resulting in improved visual and anatomical outcomes. The goal of the study was to switch patients to a treatment regimen of every 2-month injections, thereby reducing visits to long term. The final analysis of this study will be at 12 months at which time longer follow-up will help determine if the benefits of this treatment regimen are sustained over longer periods of time.
As IAI has only recently been introduced into widespread clinical use worldwide, it is highly probable that physicians will encounter subjects with active AMD who have been receiving previous anti-VEGF treatments, such as ranibizumab or bevacizumab. This study supports the transition to IAI from a PRN or TAE regimen using the fixed dosing pattern described.
Tachyphylaxis has been suggested in subjects treated for exudative AMD, and our study results might reflect this. Some eyes have shown better resolution of fluid after switching anti-VEGF treatments.17 ,24 Generally, people tend to switch subjects who are not doing well, which is defined as a lack of visual response, anatomical improvement, or both. However, whether these subjects reflect a temporal randomness in response or a true ‘treatment failure’ is difficult to determine, thus leading to selection biases especially in light of the fact that there is no standardised definition for ‘treatment failure’. Additionally, switching studies are challenging to interpret because of non-standardised intervals and protocols for evaluation, and the lack of protocol ETDRS visual acuity measurement.25 The inclusion criteria limited this study to subjects who had shown previous response to other anti-VEGF agents. Therefore, subjects were increasingly likely to respond to this or another anti-VEGF agent.
The ASSESS study tried to alleviate some of these biases; however, there were still some drawbacks to this analysis, including the small sample size with 6 months of follow-up, as well as the variable run-in to the study. It is possible that the variable lengths of time since last injection may have allowed for recurrence of fluid prior to enrolment. Prior to study entry, patients within this study had an average CST of 316 microns and an average visual acuity of 20/63. At study entry, patients had an average baseline CST of 304 microns and an average visual acuity of 20/80. These values are not significantly different, providing evidence that there may not have been significant recurrence of fluid from a possible 3-month time since last injection. Additionally, both these OCT measurements are noticeably higher than the average CST of 265.5 microns at study completion. Despite these drawbacks, the study has significant strengths including its prospective nature, strict entry criteria, and rigorous evaluation of anatomical and visual outcomes.
This study's aim was to determine the outcomes following the switch from other anti-VEGF therapies to a fixed IAI-dosing regimen. Patients were treated as per the FDA label for aflibercept which states that subjects should receive three injections monthly for the first 3 months and then every 2 months thereafter. These subjects were not necessarily ‘treatment failures’ but were more consistent with the average subjects who could experience multiple improvements and declines in their OCT. Results of this study demonstrated that 100% of subjects remained visually stable and 84.6% gained vision after switching to this dosing regimen. It is possible the subjects could have had similar improvements solely by changing to a fixed dosing pattern. However, it is our assumption that the results seen in the ASSESS study are due to a combination of exposure to a different anti-VEGF drug, with theoretical benefits over their previous treatments, and a switch to a fixed dosing regimen. The 12-month results of the ASSESS study will be helpful in further characterising these initial clinical findings.
References
Footnotes
-
Contributors RPS was responsible for: (1) substantial contributions to the conception or design of the work; the acquisition, analysis, or interpretation of data for the work; (2) drafting the work or revising it critically for important intellectual content; (3) final approval of the version to be published; (4) agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
-
Funding Support provided by a research trial grant from Regeneron Inc.
-
Competing interests None.
-
Ethics approval Cleveland Clinic Institutional Review Board.
-
Provenance and peer review Commissioned; externally peer reviewed.