Article Text

Abstract
Aim The aim of the study was to describe the clinical and genetic features of 15 Italian patients with Bietti crystalline dystrophy (BCD).
Methods All study participants underwent a complete ophthalmological examination, including standard electroretinogram (ERG), optical coherence tomography, microperimetry, autofluorescence and multifocal electroretinogram. The 11 exons of the CYP4V2 gene were sequenced. The effect of mutations on protein function was estimated by a combination of web based programs.
Results 15 patients (eight women, 7 men, aged 29–60 years) with BCD were recruited into this study. Sequencing of CYP4V2 revealed nine sequence variants in four unrelated families and six isolated individuals with BCD. Seven of these variants were novel. Among the patients, even with the same genotype, considerable variability in phenotypic expression with different degrees of accumulation of the typical intraretinal crystalline deposits was detected. Moreover, we found that more than 50% of patients had recordable standard ERG responses and in two patients the responses were within normal limits after 20 years of symptom onset.
Conclusions In conclusion, we have reported seven new mutations and illustrated the large range of genotypic and phenotypic variability in BCD, highlighting the lack of a clear genotype–phenotype correlation and underlining the existence of less severe clinical manifestations, probably linked to relatively mild mutations.
- Retina
- Genetics
- Degeneration
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Bietti crystalline dystrophy (BCD) is a rare autosomal recessive1 retinal degenerative disease, first described by Bietti in 1937.2 The disease is characterised by multiple small crystalline intraretinal deposits, associated with retinal pigment epithelium (RPE) atrophy, pigment clumping and choroidal sclerosis, with or without perilimbal subepithelial crystals.3 While BCD symptoms are limited to the visual system, crystalline inclusions are found throughout the body, including in lymphocytes and skin fibroblasts.4
Clinically, BCD is a progressive disease; affected patients experience decreased vision, night blindness and paracentral scotoma between the second and fourth decades of life. Later, patients develop peripheral visual field loss and marked visual impairment, usually progressing to legal blindness by the fifth or sixth decade of life.5
BCD is a global disease but it has been reported to be most common in East Asia, especially in Chinese and Japanese populations,6 and has been estimated to account for 3% of all non-syndromic retinitis pigmentosa cases and 10% of autosomal recessive retinitis pigmentosa cases.7
The gene responsible for BCD, CYP4V2, is expressed widely in human heart, brain, placenta, lung, liver, retina and RPE. To date, 39 mutations of CYP4V2 have been described in patients with BCD.8–13 This gene codes for a protein whose structure suggests that it may be active in fatty acid metabolism. Biochemical studies show that abnormal lipid metabolism is present in patients with BCD, and biochemical characterisation of CYP4V2 shows that it is a fatty acid omega hydroxylase.14 ,15 Histopathology shows advanced panchorioretinal atrophy, with crystals and complex lipid inclusions seen in choroidal fibroblasts, corneal keratocytes, and conjunctival and skin fibroblasts, as well as in circulating lymphocytes. These findings suggest that BCD may result from a systemic abnormality of lipid metabolism.5 ,11
Previous studies have shown a considerable heterogeneity of phenotypic expression and a good correlation between the clinical stage of disease and electroretinographic changes, showing a gradual deterioration of functional parameters in relation to the progression of retinal damage. In the early stages of the disease, the RPE–choriocapillaris complex is the first structure involved, while the functions of rods and cones are still preserved. With the extension of the atrophy of the RPE–choriocapillaris and choroidal sclerosis, there is a gradual alteration of the electroretinographic responses with progressive visual field constriction and decreased visual acuity (VA).16 ,17
The aim of the study was to describe the clinical and genetic features of 15 Italian patients with BCD and, in addition, through clinical and genetic study of the patients, to verify a possible correlation between the type of genetic mutation, phenotypic expression and disease course.
Materials and methods
Patients
This study was performed in accordance with the tenets of the Declaration of Helsinki and was approved by the internal review boards at the Second University of Naples and the National Institutes of Health CNS institutional review boards. Patients consented at the Referral Centre for Hereditary Retinopathies of the Second University of Naples. All study participants underwent a thorough medical anamnesis, genetic counselling, peripheral blood tests to assess cholesterol and triglyceride levels, and a complete ophthalmological examination, including central VA with EDTRS charts, Ishihara colour vision tests, slit lamp biomicroscopy of the anterior segment, fundus examination, fundus photography and standard electroretinogram (ERG), according to the standard protocol of the International Society for Clinical Electrophysiology of Vision.18 In addition, we performed optical coherence tomography (OCT) in 11 patients, microperimetry (MP-1) in eight patients and autofluorescence (AF) and multifocal ERG (mfERG) in six patients. BCD was diagnosed according to symptoms and typical ophthalmoscopic findings, based on the stage classification of Yuzawa et al.19
Mutation detection
Blood samples were obtained by venepuncture, and genomic DNA was extracted according to standard protocols. CYP4V2 exon and flanking sequences were determined as previously described,12 primer sequences amplifying each exon and approximately 100 bases of flanking introns. Briefly, DNA was sequenced with the ABI BigDye Terminator cycle sequencing kit v3.1, according to the manufacturer's recommendations (Applied Biosystems), through use of an ABI 3130 DNA sequencer. The sequence information was imported into Mutation Surveyor V3.25 (SoftGenetics LLC, State College, Pennsylvania, USA), and sequences of affected and normal individuals and consensus sequence were aligned to identify variations.
The effect of mutations on protein function was estimated by a combination of the SIFT,20 Polyphen,21 Mutation Assessor22 and Condel23 web based programs.
Results
Fifteen patients (eight women, seven men, aged 29–60 years) with BCD were recruited into this study. Among the patients, there were four groups of siblings (p1, p1.1, p1.2; p3, p3.1; p4, p4.1; p8, p8.1). Clinical and genetic findings of the patients are described in table 1.
Clinical and genetic data of Bietti crystalline dystrophy patients
Genetic counselling revealed parental consanguinity in only two patients (p8; p8.1) whose parents are first degree cousins. The medical history excluded systemic diseases such as cystinosis, oxalosis, gyrate atrophy and the use of drugs (tamoxifen), which can cause accumulation of intraretinal crystals.
Peripheral blood examinations showed the presence of mild hypercholesterolaemia pharmacologically untreated in five patients (p1.1, p4, p4.1, p1.2, p9) and moderate hypercholesterolaemia treated with oral lipid lowering agents in two patients (p8, p10).
Sequencing of CYP4V2 within the coding region, including intron–exon boundaries, revealed nine sequence variants in four unrelated families and six isolated individuals with BCD (table 2).
Mutations and predicted effects on CYP4V2 structure and function
Seven of these variants were detected for the first time in this study: c.364T>C (S122P), c.1393A>G (R465G), c.694C>T (R232X), c.277T>C (W93R), 724delG (_), c.1328G>A (R443Q) and c.76G>A (G26S). Two homozygous mutations were detected in one family and two isolated patients (p8, p8.1, p9, p10). Five compound heterozygous mutations were detected in three families (p1, p3, p4) and three isolated patients (p2, p5, p6) while only a single heterozygous mutation was detected in patient No 7. The R465G mutation was the most commonly detected mutation in our study population (eight of 30 alleles), although the c.332T>C (I111T) and c.694C>T (p.R232X) mutations were also seen frequently (five or 30 alleles each).
The significance of the sequence variations in terms of CYP4V2 function were predicted with the Condel program, which combines estimates from SIFT, Polyphen and Mass. Based on these criteria, the missense mutations p.R465G, p.R390H, p.W93R, p.I111T and p.R443Q are all predicted to be deleterious. For these sequence changes, there is good agreement among all programs, which is an indicator of the reliability of the change. Conversely, the novel changes p.S122P and p.G26S are predicted to be benign. The p.G26S change occurs within the transmembrane region immediately before an alpha helix, which might explain a possible effect beyond that predicted by the bioinformatic algorithms (figure 1). Similarly, the p.S122P change occurs on the end of an alpha helix, which might be disrupted by insertion of a proline at this point. In addition to the changes above, there are two mutations resulting in premature termination (p.R232X and p.D242Ifs35), both of which would be expected to result in nonsense mediated decay and essentially no protein product being produced.
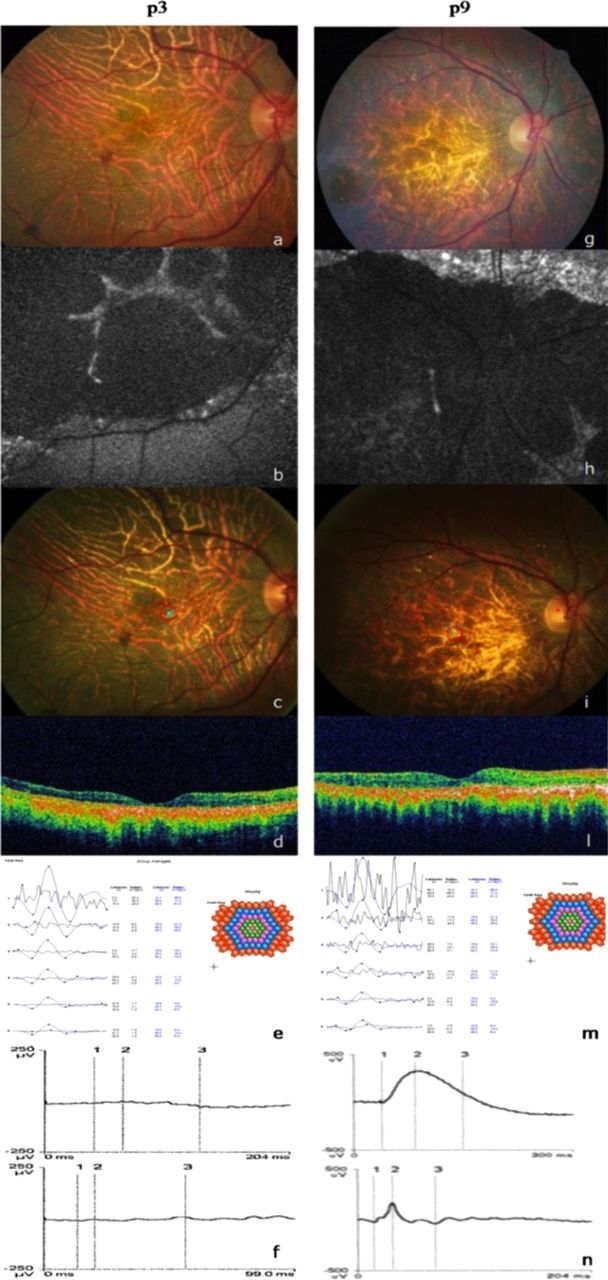
(A) Retinography, (B) autofluorescence (AF), (C) microperimetry, (D) optical coherence tomography (OCT), (E) multifocal electroretinogram (ERG), (F) standard ERG of the right eye of patient p3, (G) retinography, (H) AF, (I) microperimetry, (L) OCT, (M) multifocal ERG, (N) standard ERG of the right eye of patient p9.
In all patients, the first symptoms appeared between the third and fourth decades of life with night blindness and a progressive decrease in VA. VA ranged from light perception to 1.0. Slit lamp biomicroscopic examination showed no corneal crystals at the limbus in any of the BCD patients.
In all patients fundus examination showed the presence of intraretinal bright yellowish–white crystals disseminated in the posterior pole and mid-periphery (figure 2A, G); other retinal findings included bone spicule pigmentation, retinal vascular attenuation and diffuse choriocapillaris atrophy. Eleven patients underwent OCT examination, and the OCT showed the presence of intraretinal crystals with diffuse abnormal hyperreflectivity in the neuroretinal layers, RPE atrophy and reduction of foveal thickness (figure 2D, L). No eyes revealed a continuous inner segment/outer segment junction, while in three patients (p2, p4, p5), despite the neuroretinal disorganisation and hyperreflectivity, OCT showed a preserved external limiting membrane (ELM) in the fovea (figure 3A, B). These three patients had better VA than the other patients, with a mean value of 0.9 (±0.1 SD).

Optical coherence tomography of patient p2 (A) and p4 (B), showing a preserved external limiting membrane in the fovea.

{kind=link}
{kind=link}
{kind=link}
Model of CYP4V2 with residues showing sequence changes in affected individuals shown in space filling mode. Changed residues shaded green have Condel values, indicating that they are deleterious. Residues shaded in red have changes estimated to be neutral by Condel. Alpha helices are shown as pink ribbons, beta structures are shown as yellow ribbons, and turns and random coils are shown in blue and white.
Standard ERG analysis showed: two patients (p9, p6) (13%) with normal photopic and scotopic tracks (figure 2N); seven patients (p1, p2, p3, p5, p7, p8, p10) (47%) with subnormal photopic and scotopic tracks; one patient (p4) (7%) with microvolted photopic and extinguished scotopic values; and the remaining five patients (p1.1, p1.2, p3.1, p4.1, p8.1) (33%) with non-recordable responses (figure 2F).
MP-1 showed that three patients (p2, p3, p5) had stable fixation bilaterally (figure 2C) while the other three patients (p6, p7, p10) showed relatively unstable fixation in both eyes (figure 2I). Two patients (p.9, p. 4.1) had stable fixation in one eye and unstable fixation in the other.
mfERG analysis showed: three patients (p3, p9, p10) with bilaterally extinguished traces (figure 2E, M); one patient (p7) with central and peripheral traces extinguished in one eye and microvolted central and extinguished peripheral traces in the other. The other two patients (p4, p4.1) showed microvolted central recordings and extinguished peripheral recordings in both eyes.
AF performed on seven patients showed the presence of hypoautofluorescent areas corresponding to atrophic regions of the RPE–choriocapillaris complex in addition to hyperautofluorescent spots corresponding to the crystalline deposits (figure 2B, H).
Discussion
The examinations performed demonstrate that our patients satisfy all clinical criteria for BCD. Although none of our patients had corneal crystalline deposits at the limbus, this has also been described previously in the literature in which the crystals were found only in the retina.1 ,3 ,5 ,13 In agreement with the previous studies, 46.6% of patients showed mild to moderate hypercholesterolaemia, suggesting the possibility that BCD is derived from a systemic abnormality of lipid metabolism.5 ,15
CYP4V2 gene analysis showed a sequence variation in all patients. In three siblings (p1, p1.1, p1.2) two novel heterozygous sequence changes were detected: c.364T>C (S122P) and c.1393A>G (R465G) (table 2). The p.R465G sequence change was judged to have severe functional consequences by Polyphen, SIFT, Mass and Condel, which merges assessments by the three other programs into a single overall estimate. The R465G mutation was also detected in p3, p3.1, p4, p4.1 and p7, further supporting its identity as a causal mutation and making it one of the most common mutations detected in this study population (present in three of 10 families, or three of 20 founder alleles). The c.332T>C (p.I111T) mutation was also found in three of 10 families, but has been described in previous studies.12 ,20 This is distinctly different from studies in Asian populations, in which the c.802-8_810del17insGC mutation was the most common mutation detected.12 In p4 and her sibling, we detected the R465G mutation in one allele, a p.I111T mutation in the second allele and also a K259Q polymorphism. In p7 we could only detect a single R465G mutation in one allele. It is possible that a mutation in the other allele could be located in the promoter or in a non-coding region of the gene that we did not screen. Similarly, while we detected two changes in individuals p1, p1.1, p1.2 and p6, the p.S122P and p.G26S changes were not predicted to be deleterious by Condel and their significance is uncertain. There might also be an undetected sequence change in the promoter or other nearby regions that contributes to BCD in these patients. We detected the R232G mutation in a homozygous state in p8 and p8.1 and in a heterozygous state in p2 and p6. We detected W93R in p3 and p3.1; G26S in p6 in the heterozygous state. In p5, we detected two novel heterozygous mutations, c.724delG (p.D242Ifs35) and c.1393A>G (p.R443Q). The first is predicted to result in a frameshift of the CYP4V2 gene followed by 34 new amino acids before a termination codon. However, as the premature termination occurs in exon 6, an internal exon, it would be expected to result in nonsense mediated decay, a process by which mRNAs encoding incomplete proteins are degraded during translation.25 A similar process is expected to result from the c.694C>T (p.R232X) mutation, which also results in a premature termination in exon 6.
Among the patients, even with the same genotype, considerable variability in phenotypic expression with different degrees of accumulation of the typical intraretinal crystalline deposits was detected. In fact, p4 and her sibling, after 8 years of follow-up, showed different disease progression. Patient p4 retained good VA and recordable ERG whereas her sibling, p4.1, showed progressive VA reduction and extinguished ERG.
Moreover, three brothers (p1, p1.1 and p1.2) and brothers p8 and p8.1 also showed a high intrafamilial variability with different degrees of disease severity.
In addition, subdivision of the mutations into severe and mild, we verified if mutation severity was related to disease severity, comparing mutations to VA, ERG and fundus findings. This analysis clearly showed that there was no correlation between genotype severity (more deleterious mutations vs less destructive mutations) and clinical phenotype. For example, patient p2 presented with severe mutations but his clinical phenotype was characterised by very good VA (1/1), mild RPE dystrophy with crystalline deposits only at the posterior pole and a recordable ERG. In contrast, patient p1.1 with a mild mutation showed a severe phenotype, featuring only light perception, extensive chorioretinal atrophy and extinguished ERG.
These results suggest that there is no correlation between genotype and clinical phenotype, probably because environmental factors, such as diet, may affect lipid metabolism and hence the phenotypic expression of the disease, as mentioned in previous studies.9 ,14
Moreover, although BCD has been described as a progressive disease, age does not always correlate with severity and progression, as illustrated here. In fact, patient p1.1, 41 years old, presented with more severe morphofunctional findings compared with patient p8, aged 54 years.
All patients showed the same OCT abnormalities although three patients. p2, p4 and p5, presented with a preserved ELM layer associated with better VA, suggesting that ELM could be an important foveal photoreceptor indicator.
Furthermore, in this study, we showed that mfERG and microperimetry are more sensitive methods for follow-up of macular function in BCD patients compared with standard ERG.
In patient p9, despite the normal standard ERG responses, MP-1 showed bilateral central scotoma with relatively unstable fixation, and mfERG detected markedly depressed central responses that correlated with the extensive atrophy visible at AF and with macular thinning observed by OCT (figure 2G–N).
We found that more than 50% of patients had recordable standard ERG responses; in particular, in two patients (p.6, p9), we unexpectedly observed that the latency and amplitude of both components were within normal limits, even after 20 years of symptom onset. These data were in contrast with those reported in previous studies, in which standard ERG was correlated with disease progression.3 In these two cases, despite the thinning and impaired retinal lamination seen at sites of complete RPE/choriocapillaris atrophy, ERG responses suggest that the neural retina is still contributing to the ERG signals and is therefore still viable. This hypothesis is supported by electrophysiological studies performed in BCD patients showing that the phototransduction process can be unaffected even in advanced disease stage, with extensive RPE/choriocapillaris atrophy.16 We thought that this apparent ability of the neural retina to survive after 20 years from symptom onset is remarkable and of therapeutic and prognostic relevance, suggesting that gene or conventional therapy might remain a viable option even in the late stages of the disease.
In conclusion, in this study, we suggest that spectral domain OCT, AF, mfERG and MP-1 are useful for the diagnosis, prognosis and follow-up of BCD patients as these techniques are more detailed and more sensitive in identifying morphological and functional retinal changes that characterise disease progression. Furthermore, we have reported seven new mutations and illustrated the large range of genotypic and phenotypic variability in BCD, highlighting the lack of a clear genotype–phenotype correlation and underlining the existence of less severe clinical manifestations, probably linked to relatively mild mutations. Moreover, this study is the first to report both genetic and clinical findings of Italian BCD patients in addition to the many reports from Asia.
Acknowledgments
The authors thank Carmela Acerra for text editing.
References
Footnotes
FT and SR contributed equally to this article.
-
Contributors SR and FT conceived and designed the study; FY and CG acquired the data; SR, FT, AL and JFH analysed and interpreted the data; SR and FT drafted the article; AL, FY, CG, JFH and FS revised the manuscript critically for important intellectual content. All authors approved the final version.
-
Funding None.
-
Competing interests None.
-
Ethics approval The study was approved by the internal review boards at the Second University of Naples and the National Institutes of Health CNS institutional review boards.
Provenance and peer review Not commissioned; externally peer reviewed.
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/