Article Text

Abstract
Aim To compare the efficacy and safety of single-dose bimatoprost 0.03%/timolol 0.5% preservative-free (PF) ophthalmic solution with bimatoprost 0.03%/timolol 0.5% ophthalmic solution in patients with open-angle glaucoma or ocular hypertension.
Methods In this multicentre, randomised, parallel-group study, patients were randomised to bimatoprost/timolol PF or bimatoprost/timolol once daily in the morning for 12 weeks. Primary efficacy endpoints, reflecting differing regional regulatory requirements, included change from baseline in worse eye intraocular pressure (IOP) in the per-protocol population at week 12, and the average eye IOP at weeks 2, 6 and 12 in the intent-to-treat population.
Results 561 patients were randomised (278 to bimatoprost/timolol PF; 283 to bimatoprost/timolol); 96.3% completed the study. Both treatment groups showed statistically and clinically significant mean decreases from baseline in worse eye IOP and in average eye IOP at all follow-up time points (p<0.001). Bimatoprost/timolol PF met all pre-established criteria for non-inferiority and equivalence to bimatoprost/timolol. Ocular adverse events were similar between treatment groups, with conjunctival hyperaemia being the most frequent. Most were mild or moderate in severity.
Conclusions Bimatoprost/timolol PF demonstrated non-inferiority and equivalence in IOP lowering compared with bimatoprost/timolol, with no significant differences in safety and tolerability.
Trial registration number NCT01177098.
- Clinical Trial
- Drugs
- Glaucoma
- Treatment Medical
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Bimatoprost is a synthetic prostamide,1 which is highly effective in lowering intraocular pressure (IOP) in patients with ocular hypertension (OHT) or open-angle glaucoma.2 If single agents fail to achieve a satisfactory IOP reduction or are intolerable, fixed combinations are preferred to multiple concurrent medications.3 ,4 The fixed combination of bimatoprost 0.03%/timolol 0.5% (bimatoprost/timolol; Ganfort, Allergan, Irvine, California, USA) administered once daily has been shown to be well tolerated and to be effective in patients with inadequate IOP lowering with a single ocular hypotensive medication,5–8 as well as in treatment-naive patients.9
As topical medications are normally dispensed from multiuse bottles, preservative (typically benzalkonium chloride, BAK) is employed to maintain sterility. Although many patients use preservative-containing medications without adverse effects,10 ,11 some become sensitive to ophthalmic preservatives.12 ,13 A single-dose, preservative-free (PF) ophthalmic fixed combination would benefit this patient subpopulation. Our study evaluated the safety and efficacy of a new PF fixed-combination bimatoprost 0.03%/timolol 0.5% (bimatoprost/timolol PF; Ganfort SD) for patients who are sensitive or allergic to preservatives.
Methods
Study design and participants
This was a phase 3, multicentre, double-masked, randomised, active-controlled study (ClinicalTrials.gov: NCT01177098) conducted in 55 centres in Australia, Czech Republic, Germany, Hungary, Israel, Russia, Spain, UK and the USA. It complied with Good Clinical Practice guidelines and was approved by an institutional review board or independent ethics committee. Written informed consent was obtained from each patient prior to study enrolment.
Eligible participants were aged at least 18 years, had OHT or open-angle glaucoma, and were either treatment-naive with IOP >24 mm Hg in at least one eye or were receiving IOP treatment that was considered to be inadequate (IOP >18 mm Hg in at least one eye). At baseline, following 4 days’ to 4 weeks’ washout of IOP-lowering medications, patients were required to have an IOP of 22–30 mm Hg in each eye, with IOP asymmetry <4 mm Hg between eyes and a best-corrected visual acuity (BCVA) equivalent to a Snellen score of 20/100 or better in each eye. The minimum washout period was 4 days for parasympathomimetics and topical or systemic carbonic anhydrase inhibitors, 2 weeks for sympathomimetics and α agonists, and 4 weeks for β-adrenergic blocking agents, combination products and prostaglandin agonists. Primary exclusion criteria were: uncontrolled systemic disease; known allergy or sensitivity to the study medications or their components; introduction or anticipated alteration in ongoing use of medication that may have a significant effect on IOP; ocular surface findings (eg, hyperaemia or irritation equal to +0.5 (trace) or greater) in either eye; history (within 6 months prior to baseline) of any ocular anterior segment laser or other intraocular surgery in either eye; required chronic use of other ocular medications during the study; visual field loss that in the opinion of the investigator was functionally significant or evidence of progressive visual field loss; or anticipated wearing of contact lens in either eye during the study.
Treatment and assessments
Eligible patients were randomly assigned (1:1) at each investigator site (by automated interactive voice/web response systems) to once-daily treatment in the morning in each eye with bimatoprost/timolol PF or bimatoprost/timolol for 12 weeks. The randomisation scheme was prepared by the study sponsor. Randomisation was stratified by baseline mean diurnal IOP (≤24 mm Hg or >24 mm Hg). Patients were dispensed study medication kits containing unit-dose (single-use) containers that were identical for both formulations, thereby ensuring masking of patients, investigators and evaluators. The treatment was to be instilled in the morning to facilitate timing of study assessments. Compliance was assessed by the investigator who kept a detailed inventory of the units dispensed and reconciled containers returned to the study site.
IOP was measured using a slit lamp-mounted Goldmann applanation tonometer, and a two-person masked reading method (ie, one adjusts the dial in a masked fashion while the second reads and records the value) at 8:00 (hour 0), 10:00 and 16:00 at baseline, and at weeks 2, 6 and 12. Two consecutive measurements were taken of each eye; if these two measurements differed by ≤1 mm Hg, the IOP for the given eye was the average of the two readings. If the difference between measurements was >1 mm Hg, a third measurement was made, and the IOP for the given eye was the median of the three readings.
Safety parameters included adverse events (AEs; coded using the Medical Dictionary for Regulatory Activities V.14.1), biomicroscopy, fundus examinations (including vertical cup/disc ratio), macroscopic bulbar conjunctival hyperaemia (graded by gross inspection in comparison with standard photographs), visual acuity, visual field measurements and vital signs.
Endpoints and analyses
Two sets of primary efficacy analyses were performed: one based on worse eye IOP and one based on average eye IOP data. For worse eye IOP analysis, the efficacy endpoint was the change from baseline in worse eye IOP at week 12 in the per-protocol (PP) population (patients without a major protocol violation). Worse eye IOP referred to the eye with the higher mean diurnal IOP at the baseline visit. If both eyes had the same mean diurnal IOP at baseline, the right eye was designated as the worse eye. The treatments were compared using an analysis of covariance (ANCOVA) model with fixed effects of treatment and investigator and baseline worse eye IOP at the time-matched hour as the covariate. A two-sided 95% CI for the treatment difference (bimatoprost/timolol PF minus bimatoprost/timolol) was constructed from the ANCOVA model. Bimatoprost/timolol PF would be considered non-inferior to bimatoprost/timolol if the upper limit of the 95% CI did not exceed 1.5 mm Hg at any hour at week 12.
For the average eye analysis, IOP was evaluated at each time point (8:00, 10:00 and 16:00) at weeks 2, 6 and 12 in the intent-to-treat (ITT) population. Missing data were imputed using the last observation carried forward (LOCF) method. Treatments were compared using an analysis of variance (ANOVA) model with fixed effects of treatment and investigator. A two-sided 95% CI for the treatment difference was constructed from the ANOVA model with an estimated treatment difference based on least squares means. Bimatoprost/timolol PF would be considered equivalent to bimatoprost/timolol if the 95% CI upper limit was ≤1.5 mm Hg and the lower limit was ≥–1.5 mm Hg at all time points, and the upper limit was ≤1.0 mm Hg and the lower limit was ≥–1.0 mm Hg at the majority of time points.
The proportion of responders, defined as patients with a ≥20% reduction in worse eye IOP from the corresponding hour (hour 0, 2 or 8) of baseline at week 12, was also analysed in the ITT population. Treatment group differences were assessed using Pearson's χ2 test.
Sample size calculations
Sample size calculations were based on a one-sided α=0.025, 80% power and an assumption of no difference between treatment groups. The sample size needed to demonstrate the non-inferiority of worse eye IOP (1.5 mm Hg inferiority margin) and equivalence in average eye IOP (equivalence limit of ±1.5 mm Hg at all follow-up time points, and of ±1.0 mm Hg at the majority of follow-up time points) were estimated. The largest sample size from these estimates ensured adequate power for all criteria, given an expected 10% dropout rate.
Results
Baseline demographics and patient characteristics
The study was initiated on 31 October 2010 and completed on 21 February 2012. A total of 561 patients were enrolled with 540 (96.3%) completing the study; 278 were randomised to bimatoprost/timolol PF and 283 were randomised to bimatoprost/timolol (see online supplementary figure S1). The demographic and baseline data did not show any significant differences between the groups (table 1).
Demographics and baseline characteristics (intent-to-treat population)
Efficacy
Worse eye PP analysis
At baseline, there were no statistically or clinically significant differences in mean worse eye IOP between bimatoprost/timolol PF and bimatoprost/timolol in the PP population. Mean baseline worse eye IOP at 8:00, 10:00 and 16:00, respectively, was 25.4, 24.8 and 23.9 mm Hg in the bimatoprost/timolol PF group and 25.4, 24.7 and 23.8 mm Hg in the bimatoprost/timolol group (figure 1). Both treatment groups showed a statistically significant mean decrease from baseline in worse eye IOP at all follow-up time points (p<0.001), with mean changes from baseline IOP ranging from –9.16 to –7.98 mm Hg for the bimatoprost/timolol PF group, and from –9.03 to –7.72 for the bimatoprost/timolol group across the 12-week study.

Mean (±SD) worse eye IOP at each time point through 12 weeks of study in the per-protocol population. IOP, intraocular pressure; PF, preservative-free.
Bimatoprost/timolol PF met the criteria for non-inferiority to bimatoprost/timolol with respect to change from baseline worse eye IOP at each hour evaluated (8:00, 10:00 and 16:00) at week 12 in the PP population (figure 2). The upper limit of the 95% CI of the between-group difference did not exceed 0.14 mm Hg at week 12 and was well within the 1.5 mm Hg non-inferiority margin at each hour. The mean difference between treatment groups in change from baseline in worse eye IOP ranged from −0.37 to –0.30 mm Hg at week 12, favouring bimatoprost/timolol PF.

Treatment differences (bimatoprost/timolol PF−bimatoprost/timolol) at each time point in change from baseline worse eye IOP in the per-protocol population. IOP, intraocular pressure; PF, preservative-free. Adapted from Day et al.14
Responder ITT worse eye analysis
The percentage of patients achieving at least a 20% reduction in worse eye IOP at week 12 ranged from 86.3% to 90.6% for the bimatoprost/timolol PF group, and 85.5% to 89.8% for the bimatoprost/timolol group (ITT population; table 2). There were no statistically significant differences between the treatment groups.
Treatment comparison of responders, intent-to-treat population*
Average eye ITT analysis
There were no differences in average eye IOP between the bimatoprost/timolol PF and bimatoprost/timolol groups in the ITT population at any time point (LOCF). Both treatments showed statistically significant mean decreases from baseline in average eye IOP at all follow-up time points (p<0.001). Across the 12-week study, these changes ranged from –8.72 to −7.57 mm Hg for the bimatoprost/timolol PF group, and from −8.55 to −7.27 mm Hg for the bimatoprost/timolol group.
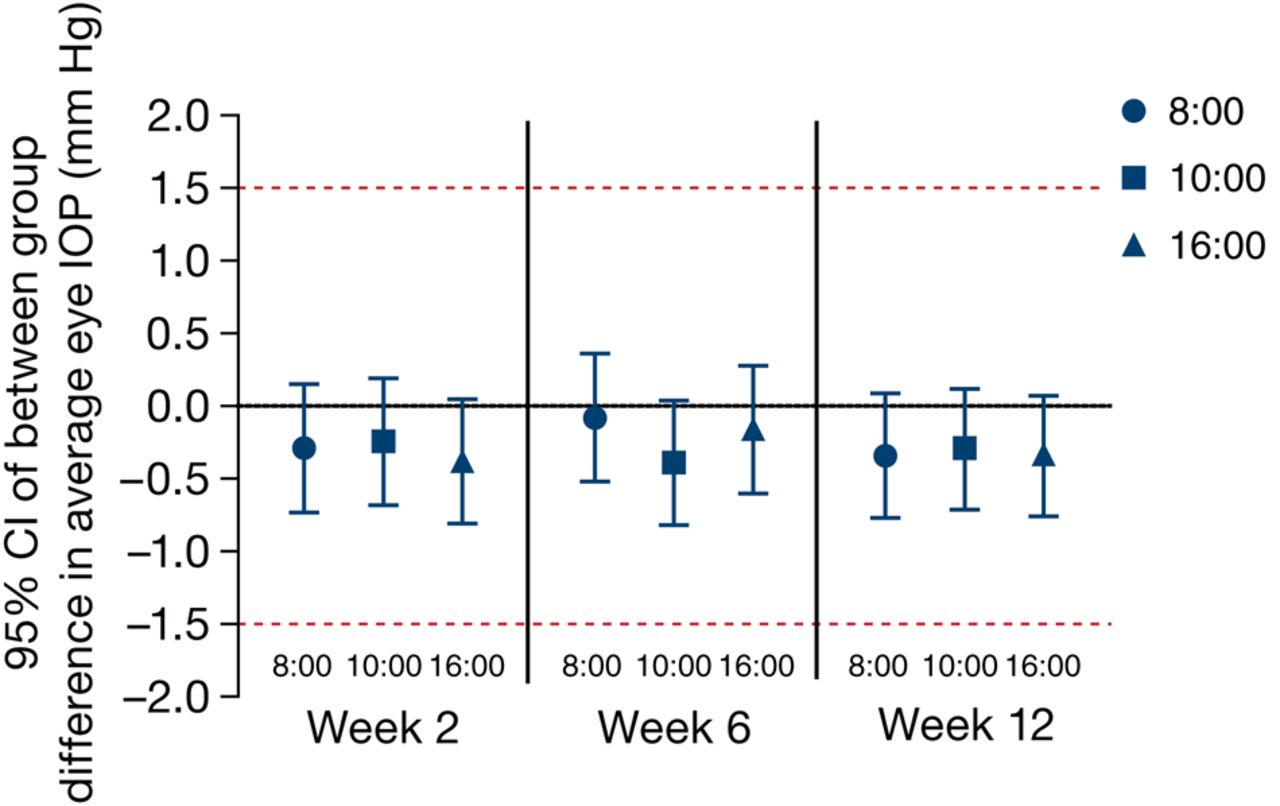
Bimatoprost/timolol PF met the criteria for equivalence to bimatoprost/timolol (figure 3). The upper limit of the 95% CI of the between-treatment difference in average eye IOP was ≤1.0 mm Hg, and the lower limit was ≥–1.0 mm Hg at all follow-up time points. At no time point was the lower limit of the 95% CI less than –0.82 mm Hg, nor did the upper limit of the 95% CI exceed 0.36 mm Hg. The treatment difference in average eye IOP ranged from –0.39 to –0.08 mm Hg at weeks 2–12, favouring bimatoprost/timolol PF.
{kind=link}
{kind=link}
{kind=link}
Treatment differences (bimatoprost/timolol PF−bimatoprost/timolol) at each time point in average eye IOP (intent-to-treat population). Missing values were imputed using the last observation carried forward method. IOP, intraocular pressure; PF, preservative-free.
Safety and tolerability
Both treatments were well tolerated. Ocular AEs were reported for 33.1% (92/278) of patients in the bimatoprost/timolol PF group and 33.7% (95/282) in the bimatoprost/timolol group and were similar in the two groups, with no significant difference in overall frequency (p=0.881) or frequency of any individual event (p≥0.063; table 3). The most frequent ocular AE in each group was conjunctival hyperaemia, which was usually mild in severity. Treatment-related AEs were reported in 28.8% (80/278) and 28.7% (81/282) of patients in the bimatoprost/timolol PF and bimatoprost/timolol groups, respectively. The most common treatment-related AEs, occurring in ≥2% of patients, were conjunctival hyperaemia, eye pruritus, skin hyperpigmentation, dry eye, eye pain, growth of eyelashes and eyelid erythema. The only treatment-related AE with a statistically significant difference in incidence between treatment groups was skin hyperpigmentation (4.0% (11/278) and 1.1% (3/282) in the bimatoprost/timolol PF and bimatoprost/timolol groups, respectively; p=0.028).
Ocular adverse events reported in ≥2.0% of patients in either group during the 12-week study period (safety population), n (%)
Eight serious AEs (four in the bimatoprost/timolol PF group and four in the bimatoprost/timolol group) were reported, including one death due to a metastatic pulmonary malignancy in the bimatoprost/timolol group deemed by the investigator as unrelated to the study drug or procedure (see online supplementary table S1). The diagnosis of malignancy was made 2 months after randomisation. In all, 13 discontinuations caused by AEs (four in the bimatoprost/timolol PF group and nine in the bimatoprost/timolol group) were reported, with the majority being ocular AEs (see online supplementary figure S1).
On biomicroscopy, the percentage of patients with a ≥1 severity grade increase in conjunctival hyperaemia from baseline was 18.0% (50/278) in the bimatoprost/timolol PF group, and 17.0% (48/282) in the bimatoprost/timolol group (p=0.764). On biomicroscopic and macroscopic assessments, severity grades of hyperaemia were similar between the bimatoprost/timolol PF group and the bimatoprost/timolol group (see online supplementary figure S2). The overall between-group difference was p=0.943 for biomicroscopy evaluation of conjunctival hyperaemia and p=0.852 for macroscopic evaluation of bulbar hyperaemia. Most cases were trace to mild, with just two cases classified as severe (in the bimatoprost/timolol group, on biomicroscopic and macroscopic evaluations).
Finally, no statistically or clinically significant between-group differences were noted for change from baseline in cup/disc ratio, BCVA, or visual field loss at week 12. At the final evaluation, 98.9% of patients had no change (between >−0.2 and <+0.2) in cup/disc ratio from baseline; three patients in each group had worsening (defined as a positive change of ≥0.2). Likewise, a worsening of BCVA (defined as a decrease of ≥2 lines) was detected in 6.3% (17/278) of patients in the bimatoprost/timolol PF group, and 5.6% (15/282) in the bimatoprost/timolol group (p=0.25). On the visual field assessment, 13 patients in the bimatoprost/timolol PF group shifted from normal at baseline to abnormal at week 12, whereas 15 patients shifted from abnormal to normal. In the bimatoprost/timolol group, 16 patients shifted from normal at baseline to abnormal at week 12, and 11 patients shifted from abnormal at baseline to normal at week 12. These findings were not clinically significant. Similarly, there were no statistically or clinically significant differences between groups in vital signs.
Discussion
In the treatment of patients with OHT or glaucoma, IOP lowering preserves visual function.15–19 Evaluation of data from randomised controlled glaucoma trials have shown significant reductions in the risk of progression with decreases in IOP.16 ,19 Although the first-line option for lowering IOP is typically a single hypotensive agent, patients frequently require a second IOP-lowering agent. A fixed-combination preparation is preferable to consecutive separate instillations of the two agents, in that it may improve adherence by decreasing the number of drops needed.3 ,4 This may be particularly relevant in patients with ocular surface disease possibly aggravated by preservatives,20 ,21 and for whom multiple instillations of eye drops may exacerbate associated difficulties.
In a multicentre, observational, open-label study of patients with inadequate IOP lowering, bimatoprost/timolol was found to reduce IOP with improved or equivalent tolerability, compared with prior medications, with good adherence to treatment.22 Reductions in hyperaemia and IOP were statistically significant when patients were switched from bimatoprost, latanoprost or travoprost monotherapy to a fixed combination of bimatoprost/timolol in an open-label longitudinal study.23 Fixed-combination bimatoprost/timolol has demonstrated better IOP-lowering efficacy than latanoprost/timolol in a randomised crossover study,24 as well as better IOP-lowering efficacy than travoprost/timolol in a crossover comparison study in patients previously treated with latanoprost/timolol fixed combination.25 Similarly, in a meta-analysis of 20 clinical trials comparing fixed combinations of prostaglandin analog/timolol with prostaglandin agonist monotherapies, bimatoprost/timolol was found to provide significantly greater IOP reduction than latanoprost-based and travoprost-based combinations, as well as bimatoprost alone (in most studies).26 A more recent meta-analysis of five trials comparing fixed and unfixed timolol combinations found fixed combinations of the three prostaglandin agonists to be less effective than concurrently administered agents in an unfixed combination (p=0.0007); the studies, however, were characterised by significant quantitative heterogeneity (I2=58%),27 suggesting that the observed effect was likely due to inconsistency between studies.28
Patients with a known sensitivity would benefit from PF ophthalmic formulations.12 ,13 This study compared a PF formulation of bimatoprost/timolol with the standard BAK formulation. The principal conclusion is that bimatoprost/timolol PF demonstrated non-inferiority and equivalence in IOP lowering compared with bimatoprost/timolol, with differences between the treatments in IOP lowering consistently favouring bimatoprost/timolol PF. There were no significant differences in safety and tolerability between the two formulations except for treatment-related skin pigmentation which was reported more frequently in patients receiving bimatoprost/timolol PF. Differences in reports of skin hyperpigmentation overall were not statistically significantly different between groups. Based on ex vivo pharmacokinetic data, increasing BAK concentration should enhance penetration of bimatoprost across human skin (data on file, Allergan), and the incidence of skin pigmentation would be expected to be greater with bimatoprost/timolol than the PF formulation; the opposite was observed in our study. Therefore, the higher incidence of skin hyperpigmentation with bimatoprost/timolol PF is likely an incidental finding.
In conclusion, bimatoprost/timolol PF provided an effective IOP-lowering alternative to bimatoprost/timolol for patients who are not sufficiently controlled on monotherapy and/or are sensitive to preservatives. Studies that address the present design limitations are warranted, such as including patients with ocular surface disease and those using multiple topical medications over a longer duration, analysing patients who required additional therapy during long-term use, and cost effectiveness of the treatment options based on individual patient needs.
Acknowledgments
Medical writing assistance was provided by Linda Whetter, DVM, PhD, and Diann Glickman, PharmD, of Evidence Scientific Solutions and was funded by Allergan, Inc. Participating Investigators: The authors wish to thank the investigators of the Ganfort Preservative-free Study for allowing their patients to be included in this trial. Australia: Ashish Agar, Michael Coote, Ivan Goldberg. Czech Republic: Igor Vicha, Zdenka Wandrolová. Germany: Julia Clauss, Hanna Ettinger-Neuss, Joachim Götting, Sabine Huhle, Wolfram Lieschke, Ellen Machnik, Norma Nenning, Michael Vobig. Hungary: András Berta, Zsolt Biró, Gábor Holló, Lajos Kolozsvári, Pál Sziklai. Israel: Orna Geyer, Shimon Kurtz, Moshe Lusky, Shlomo Melamed, Lilly Naveh, Zvi Segal, Miriam Zalish. Russia: Yuri Astakhov, Valeriy Erichev. Spain: Aitor Lanzagorta Aresti, Rafael Gil Pina, Mercé Guarro, María Isabel Canut Jordana, Vicente Polo, José Manuel Navero Rodríquez, José Isidro Belda Sanchis, Fernando Ussa Herrera. UK: Rupert Bourne, Francesca Cordeiro, K Sheng Lim. USA: Louis Alpern, William Christie, Efraim Duzman, Richard Evans, Robert Foerster, Raymond Fong, Barry Katzman, Thomas Macejko, Joseph Peters, Steven Rauchman, Kenneth Sall, Gail Schwartz, Steven Simmons, Gregory Sulkowski, Thomas Walters, David Wirta.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors IG and MB are guarantors for this paper. IG, RMS and MB participated in the study design, data interpretation, and drafting, critical review, revision, and approval of the manuscript. RGP and AL-A participated in the data collection, and critical review, revision, and approval of the manuscript. CL conducted the data analysis, and critically reviewed, and approved the manuscript. The study sponsor participated in the study design, analysis and interpretation of the data, in writing the report and in the decision to submit the paper for publication.
-
Funding The study and medical writing support were sponsored by Allergan, Inc, Irvine, California, USA.
-
Competing interests IG is consultant for Allergan, Alcon Laboratories, Forsight, Merck and Pfizer, an advisory board member for Allergan, Alcon Laboratories, Merck and Pfizer, and a speaker for Allergan, Alcon, and Pfizer. RGP and ALA have no financial disclosures to declare. RMS, CL and MB are employees of Allergan, Inc. RMS holds a patent relevant to the work.
-
Ethics approval As this was a multicenter study, several IECs and IRBs approved the study.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement For additional unpublished data from the study, Allergan should be contacted.