Article Text

Abstract
Background/Aims Prospective data on switching anti-vascular endothelial growth factors in patients with neovascular age-related macular degeneration (nAMD) who have previously shown no/partial response are limited. This prospective study assessed the effect of switching from aflibercept to ranibizumab on anatomical and functional outcomes in patients with persistent/recurrent disease activity.
Methods SAFARI (NCT02161575) was a 6-month, prospective, single-arm study conducted in the UK and Germany. Patients, meeting strict eligibility criteria for one of two subgroups (primary treatment failure or suboptimal treatment response), received 3 monthly intravitreal ranibizumab injections (0.5 mg). Thereafter, ranibizumab was administered pro re nata at monthly visits. The primary endpoint was change from baseline (CfB) to day 90 in central subfield retinal thickness (CSRT). Best-corrected visual acuity (BCVA) and retinal morphology parameters were assessed.
Results One hundred patients were enrolled (primary treatment failure, 1; suboptimal treatment response, 99). In the overall population, there was a significant CfB in median CSRT of −30.75 µm (95% CI −59.50,–20.50; p<0.0001) to day 90. Improvements were also observed in other quantitative and qualitative optical coherence tomography parameters. In Early Treatment Diabetic Retinopathy Study letters assessed by category, 55% and 59% of patients gained 0–≥15 letters versus baseline at day 90 and day 180, respectively. However, mean improvements in BCVA (CfB) to each time point were small (≤2 letters). No new safety signals were identified.
Conclusion Switching from aflibercept to ranibizumab led to a significant improvement in CSRT, with ~60% experiencing stabilised/improved BCVA. Therefore, patients with nAMD who have shown a suboptimal response to aflibercept may benefit from switching to ranibizumab.
- clinical trial
- degeneration
- imaging
- macula
- neovascularisation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Age-related macular degeneration (AMD) is the leading cause of severe vision loss in people aged ≥50 years in the UK and other European countries.1–3 In both the developed and developing world, the burden of this disease is projected to increase due to ageing populations and rising life expectancies, respectively.3–5 The wet or neovascular form of AMD (nAMD) accounts for most cases associated with severe visual impairment.6
Intravitreal (IVT) vascular endothelial growth factor-A (VEGF-A) inhibitors (anti-VEGFs), such as ranibizumab and aflibercept, are now the standard of care.7 8 Ranibizumab has demonstrated clinically and statistically significant improvements in visual acuity (VA) and quality of life for patients with nAMD.9 Aflibercept was subsequently demonstrated to be clinically equivalent to ranibizumab with respect to improvement in VA at 52 weeks.10
Despite anti-VEGF therapy, a minority of patients still experience rapid, progressive deterioration in clinical status, with no demonstration of an adequate clinical response (primary treatment failure). This group contains patients who will not respond to continued therapy with the same anti-VEGF, termed ‘non-responders’. A further subgroup experience persistent or recurrent disease activity following an initial response to therapy (suboptimal treatment response).11 12 While anti-VEGF resistance is a well-established phenomenon, its causes remain poorly understood.13 An occasionally accompanying mechanism is tachyphylaxis, describing a reduction in response to a drug, often occurring following repeat administration.13–15
In the clinical setting, if an inadequate or diminishing response is observed, increasing dosing frequency to monthly injections until visual and anatomical stability is achieved, or switching to an alternative anti-VEGF may be considered. However, limited prospective data exist on retinal morphology and other potential benefits and risks associated with switching between anti-VEGFs in nAMD. Data on switching from aflibercept to ranibizumab are particularly sparse, with the majority being derived from ‘switch-back’ studies.16 17
This study determined whether switching treatment to ranibizumab reduces disease activity in patients with nAMD treated with aflibercept and in whom there was evidence of persistent or recurrent disease activity. Furthermore, this study evaluated retinal morphology to identify potential predictors of response to ranibizumab.
Methods
Patients
Eligible patients were aged ≥50 years, with prior aflibercept treatment meeting strict criteria (Box 1) and with best-corrected visual acuity (BCVA) ≥23 Early Treatment Diabetic Retinopathy Study (ETDRS) letters in the study eye, at both screening and baseline. The patients had an active, angiographically documented, choroidal neovascularisation (CNV) lesion secondary to AMD in the study eye at screening, and evidence of active CNV in the centre of the study eye fovea at baseline. They also had a total area of fibrosis in the study eye comprising <50% of the lesion area, as assessed by all imaging data available, including fluorescein angiography, fundus photography and fundus autofluorescence.
Subgroup-specific patient inclusion criteria
1. Primary treatment failure
No prior anti-VEGF treatment
Received no more than 3 injections of aflibercept into study eye prior to screening
Historical OCT volume scan acquired ≤28 days before first aflibercept injection was administered to study eye
Initiated treatment with aflibercept <130 days prior to the screening visit
Last injection of aflibercept was ≥28 days but ≤40 days prior to the baseline visit
No increase in BCVA (≥5 letters) since commencing treatment with aflibercept
Disease activity had never been controlled in the study eye after initiating aflibercept as defined by at least one of the following:
Evidence of unchanged or increasing retinal* or subretinal fluid
New PED
Unchanged or increasing size of pre-existing PED
2. Suboptimal treatment response
No prior anti-VEGF treatment
Aflibercept commenced ≥6 months prior to the screening visit
Received ≥3 aflibercept injections into the study eye within 6 months of the screening visit
Historical OCT volume scan acquired ≤28 days before first aflibercept injection was administered to study eye
Evidence of previous reduced disease activity (as defined by reduction of ≥50 µm in CSRT on OCT) noted in the study eye after initiating aflibercept
Last injection of aflibercept was ≥28 days but ≤70 days prior to the baseline visit
At screening, disease activity has worsened, as defined by increasing retinal* or subretinal fluid, or new or increasing size of PED in the study eye compared with prior visits
*Evidence of increasing retinal fluid included increased number, size or total volume of IRCs, or increased central retinal or foveal thickness, or similar quantitative retinal imaging data recorded within the individual patient record.
BCVA, best-corrected visual acuity; CSRT, central subfield retinal thickness; ETDRS, Early Treatment Diabetic Retinopathy Study; IRC, intraretinal cyst; OCT, optical coherence tomography; PED, pigment epithelial detachment; VEGF, vascular endothelial growth factor.
In addition, all patients had to meet the criteria for one of two groups: primary treatment failure or suboptimal treatment response (Box 1; online supplementary table 1).
Supplemental material
Study design
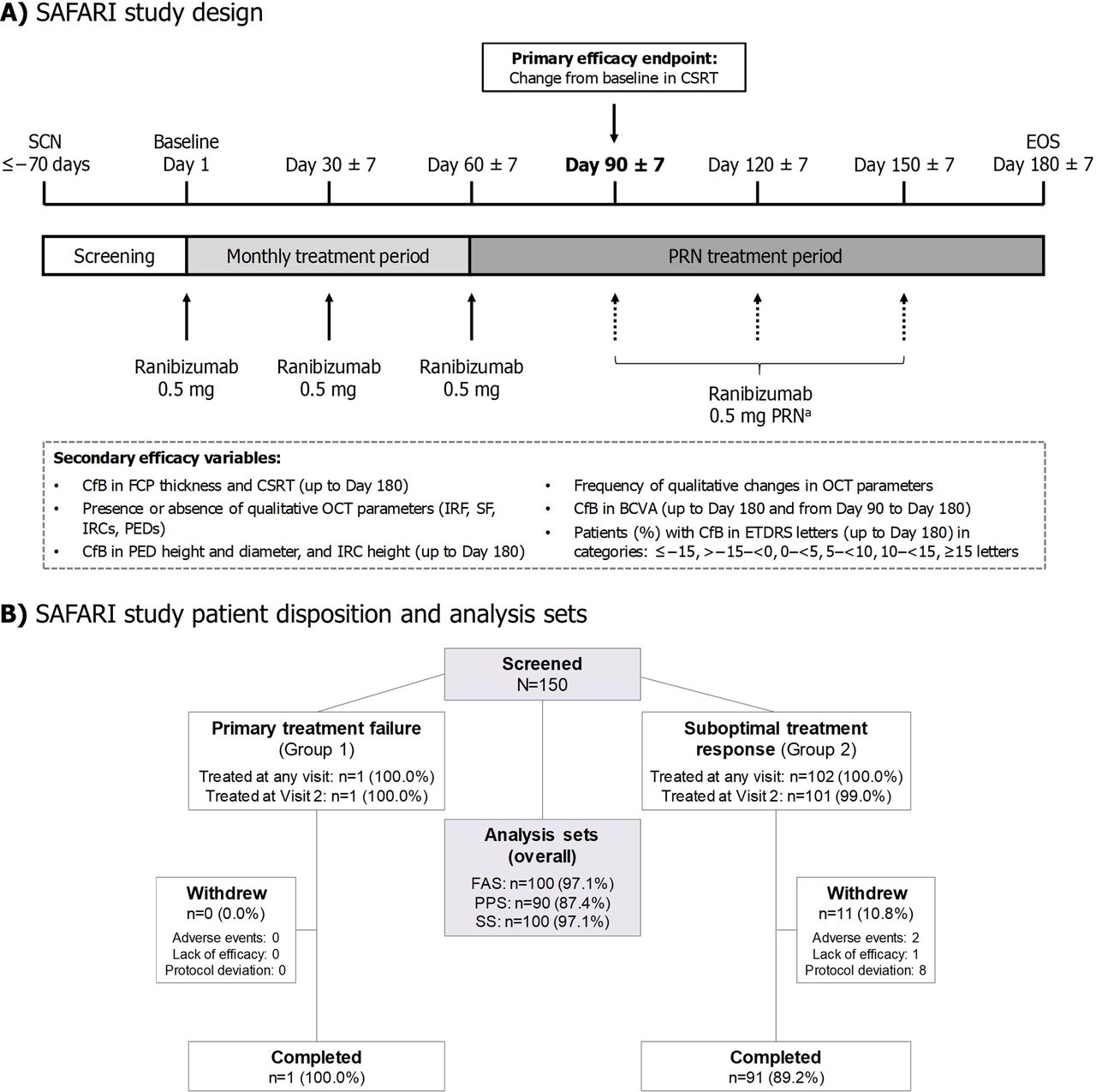
SAFARI (NCT02161575) was a 6-month, phase IV, prospective, single-arm, multicentre study. Ranibizumab (0.5 mg) was administered by monthly IVT injection on day 1 (baseline), day 30 (±7 days) and day 60 (±7 days) (figure 1A). Thereafter, patients received IVT ranibizumab (0.5 mg) on a pro re nata (PRN) basis, at the investigators’ discretion, at monthly study visits (±7 days): day 90, day 120 and day 150. An end-of-study (EOS) visit was scheduled for day 180 (±7 days).

Safari study design, patient disposition and analysis sets. Percentages=n/(patients treated at any visit)×100. A total of 10 patients were excluded from the PPS (overall) based on protocol deviations. aThe PRN regimen permitted retreatment with ranibizumab if, in the opinion of the investigator, persistent or worsening visual symptoms were attributable to nAMD, and/or if there was evidence of worsening visual acuity determined by ETDRS BCVA (>1 letter decline since last study visit), and/or the presence of persistent or worsening disease activity on OCT (eg, presence of subretinal fluid, persistent or increased number, size, or total volume of IRC, or increased central retinal or foveal thickness). BCVA, best-corrected visual acuity; CfB, change from baseline; CSRT, central subfield retinal thickness; EOS, end of study; ETDRS, Early Treatment Diabetic Retinopathy Study; FAS, full analysis set; FCP, foveal centre point; IRC, intraretinal cyst; IRF, intraretinal fluid; nAMD, neovascular age-related macular degeneration; PED, pigment epithelial detachment; PPS, per protocol set; OCT, optical coherence tomography; PRN, as required (pro re nata); SCN, screening; SF, subretinal fluid; SS, safety set.
The study was conducted from August 2014 to September 2017 at 22 study sites in the UK and 6 sites in Germany.
Efficacy and safety assessments
The primary efficacy variable was change from baseline (CfB) to day 90 in central subfield retinal thickness (CSRT), defined as the average retinal thickness (μm) of the circular area within 1 mm diameter around the foveal centre. The spectral domain-optical coherence tomography (SD-OCT) instruments that were permitted are described in the online supplementary materials.
The secondary efficacy variables are presented in figure 1A. BCVA was assessed with refraction on ETDRS charts. Colour fundus photography, fluorescein angiography and fundus autofluorescence imaging were conducted at screening and at the EOS. SD-OCT was acquired at each visit, including at screening. Indocyanine green angiography was performed at screening only. All imaging data were independently reviewed by a central reading centre (CRC, GRADE Reading Centre, Bonn, Germany) to ensure standardised evaluation.
The safety assessments included adverse events (AEs), ophthalmic examinations and vital signs. AEs were coded using MedDRA, V.20.1.
Exploratory analysis
The exploratory variables presented include CfB in subfoveal choroidal (SFC) thickness to day 180, CfB in area of macular CNV lesion, change in area of leakage from screening to day 180, presence of dry retina and the correlation between change over time in CSRT or BCVA and baseline parameters, as well as prior patient treatment history. The regression analysis methodology for assessing the effects of baseline parameters on change in CSRT and BCVA is presented in the online supplementary materials.
Statistical analysis
Sample size and power analyses, summary statistics and analysis set definitions are outlined in the online supplementary materials.
Primary efficacy assessment
Due to the non-normal distribution of the observed data for the primary efficacy variable (CSRT), the prespecified, Wilcoxon signed-rank test was used. A sensitivity analysis verified the use of the Wilcoxon signed-rank test; a transformation of ln((−1*CSRT change)+100) was applied to transform the data to a normal distribution, and a paired t-test was carried out on the transformed data. Where CSRT data were missing for day 90, the analysis was performed using a last-observation-carried-forward approach.
Secondary and exploratory efficacy assessments
There was no replacement of missing data for the secondary or exploratory variables. For summary statistics presenting percentages, the denominator is the overall N value (ie, for full analysis set (FAS), N=100) and not the number of patients at each time point.
Results
Patients
Despite two extensions to the recruitment period due to slow enrolment, recruitment was halted before the planned sample size of 124 patients had been reached. Nevertheless, a sample size of 102 had 83% power to detect a change of 30 µm in CSRT to day 90 (primary efficacy variable).
A total of 103 patients received at least one dose of study drug; of these, 92 (89.3%) completed the study, and 11 (10.7%) discontinued prematurely (figure 1B). The number of patients included in each analysis set is summarised in figure 1B. The mean (SD) age of the 100 patients in the FAS was 77.0 (6.5) years (table 1). The patients were predominantly Caucasian (99%; Asian, 1%) with a similar proportion of males and females (45% and 55%, respectively).
Patient demographics, baseline characteristics and treatment history
The patients’ treatment history prior to the switch is presented in table 1.
A median of 6.0 ranibizumab injections (range 1–6; N=100 (safety set)) were administered in the study eye. The decision to initiate treatment during the PRN phase was predominantly based on anatomical (OCT) data.
Primary efficacy variable
There was a statistically significant median decrease in CSRT of −30.75 µm (95% CI −59.50,–20.50; p<0.0001) from baseline to day 90 (figure 2A–B; online supplementary table 2). Further analysis using the per protocol set demonstrated a statistically significant median decrease of −29.25 µm (95% CI −58.50,–19.00; p<0.0001).

Changes in quantitative retinal morphology and qualitative OCT parameters with ranibizumab treatment up to 6 months (N=100). Full analysis set. aThe primary efficacy endpoint was CFB in CSRT to day 90; ***p<0.0001. Data presented in A and B represent median±min/max (as not normally distributed) and mean±SD, respectively. Baseline was defined as the last available non-missing value collected just prior to the start of treatment in the study eye. Visit days represent day ±7 days. BL, baseline; CfB, change from baseline; CSRT, central subfield retinal thickness; FCP, foveal centre point; NA, not accessible; OCT, optical coherence tomography.
Secondary efficacy variables
Decreases in median (or mean) CSRT and foveal centre point (FCP) thickness were observed throughout the monthly and PRN treatment periods (figure 2A–B; online supplementary table 2). The maximum median CfB achieved in CSRT and FCP thickness occurred at day 30 and were −35.00 µm and −38.00 µm, respectively. Maximum pigment epithelial detachment (PED) and intraretinal cyst (IRC) height were also reduced with treatment; the maximum median CfB, −24.25 µm and −33.50 µm for maximum PED and IRC height, respectively, were recorded at day 60 (online supplementary table 3). Improvements in qualitative OCT parameters (intraretinal fluid, subretinal fluid, IRCs and PEDs) are illustrated in figure 2C–F.
Mean gains in BCVA in the study eye from baseline to each time point were generally small (≤2 letters) (online supplementary table 4). No change in BCVA was observed in the study eye from day 90 to day 180 (online supplementary table 4).
There was some evidence of improvement in ETDRS letters, when assessed by category. At day 90, 29%, 14%, 7% and 5% of patients had gained 0–<5, 5–<10, 10–<15 and≥15 letters versus baseline, respectively. At day 180, 25%, 17%, 6% and 11% had gained 0–<5, 5–<10, 10–<15 and≥15 letters compared with baseline, respectively (figure 3 and online supplementary file 1).

{kind=link}
{kind=link}
{kind=link}
Change from baseline in best-corrected visual acuity (letters) (N=100). Full analysis set. aData for 1 patient are missing at day 30. Visit days represent day ±7 days.
Safety variables
Overall, 11 patients (11.0%) experienced an ocular treatment-emergent AE (TEAE) that was suspected to be related to study drug treatment by the investigator (online supplementary table 5). Most of these events were mild, and all resolved; none were severe. TEAEs leading to study discontinuation were reported by two patients (2.0%); these were the treatment-emergent serious adverse events (SAEs) of transient ischaemic attack (TIA) and retinal haemorrhage. The case of TIA, which occurred on day 150 and was mild, was the only SAE considered to be related to the study drug treatment by the investigator. Concomitant medication/therapy was administered, and the event resolved on the same day. No patients had a TEAE leading to death.
Exploratory efficacy variables
Overall, no marked CfB to day 180 was observed in SFC thickness, or from screening to day 180 in area of leakage. Small numerical changes were documented from screening to day 180 in the exploratory variables of area of total lesion and macular CNV (online supplementary table 6). No patients had dry retina during the study.
Regression analyses demonstrated that greater baseline area of leakage and FCP thickness were associated with reduced CSRT to day 90, whereas higher maximum PED diameter and baseline BCVA were significantly associated with an increase in CSRT to day 90, after adjusting for baseline risk factors (online supplementary results).
Discussion
The SAFARI study assessed the anatomical and visual outcomes of switching from aflibercept to ranibizumab following persistent or recurrent disease activity in patients with nAMD. The primary endpoint showed a statistically significant median CfB of −30.75 µm in CSRT to 3 months, following monthly IVT injections with ranibizumab. There was also evidence of improvement after switching in both quantitative and qualitative OCT parameters, and ETDRS letters assessed by category. While the changes observed were relatively small, the results demonstrated OCT stability following the switch to ranibizumab, with stabilised or improved BCVA for ~60% of this challenging-to-treat patient population. No new safety findings were identified.18
Collectively, anti-VEGF switching studies have suggested clinical benefit following switching for poor responders with nAMD, typically reporting a small gain in ETDRS letters.17 19 However, these findings are largely derived from retrospective and uncontrolled studies with heterogeneous criteria for ‘poor responders’. The SAFARI study is the first prospective study to have examined the potential benefits and risks associated with anti-VEGF switching in patients with nAMD with a poor response to aflibercept treatment meeting strict criteria.
The putative mechanisms underlying anti-VEGF resistance and considerations regarding classification of ‘responder status’ have been discussed elsewhere.13–15 17 There is no consensus on the classification of ‘non-responders’ or ‘poor responders’, and the defining criteria employed in the literature vary.13 In this study, patients were eligible for inclusion in the suboptimal response group if they had worsened disease activity at screening (increasing retinal or subretinal fluid, or new or increasing in size PED in the study eye compared with prior visits).
In this 6-month study, 59% of patients switching to ranibizumab following a suboptimal response with aflibercept experienced stabilised or improved BCVA to day 180, although this was counterbalanced by a large minority (38%) who lost ≥1 letter. The lack of a substantial improvement in mean VA requires further investigation, but it may be due, in part, to the relatively long duration of disease activity prior to the switch (mean±SD total duration of prior aflibercept treatment: 325.5 days±160 (min: 63; max: 933 days)), which could have resulted in some of the structural changes becoming permanent. Switching early on may be helpful in elucidating the most appropriate drug and regimen for individual patients to preserve vision in the long term. However, this study did not ultimately assess the impact of switching early, as only one patient was recruited to the primary treatment failure group, and further research on this is required.
The largest improvements in clinical outcomes in the SAFARI study were observed during the initial monthly treatment period. This finding was anticipated given that these patients were experiencing persistent or recurrent disease activity with aflibercept before being switched to a monthly treatment regimen with ranibizumab. In the PRN phase, there was some evidence of the CSRT response diminishing; however, the difference between the median CfB at day 90 and day 180 was only 1.25 µm, which is likely to be of minimal clinical significance. While the clinical significance of this statistically significant reduction in CSRT to day 90 is unclear, it is supported by a trend towards improved BCVA. Regression analyses demonstrated that baseline parameters such as greater area of leakage and FCP thickness, and higher maximum PED diameter and BCVA, were associated with reduced or increased CSRT to day 90, respectively. The value of these parameters as potential predictors of response to ranibizumab merits further investigation.
In addition to switching anti-VEGFs, the importance of regular dosing cannot be understated. The lack of a tightly controlled monthly treatment regimen is thought to account for the poor translatability of VA gains associated with clinical trials in the real-world setting.19–22 It is also possible for patients to experience similar clinical improvements to those achieved by switching anti-VEGF by simply reverting to fixed dosing with their current anti-VEGF treatment.23 In line with this, in patients with nAMD who required monthly treatment following a 2-year trial, no significant difference in clinical outcomes was reported between continuing on ranibizumab and switching to aflibercept.24 This is also reflected in clinical practice, where if the treatment response achieved is inadequate or diminishes, the strategy is often to increase the dosing frequency of the current anti-VEGF to monthly injections. Thus, prior underdosing with the first anti-VEGF therapy, by transitioning from a ‘real life’ paradigm to a prospective study protocol, can be a confounding factor. However, a key strength of the SAFARI study was that patients had to meet strict inclusion criteria with respect to prior aflibercept treatment, including the number and timeframe of injections.
At the time of study in the UK, licensed treatment with aflibercept in year 1 consisted of 3 monthly IVT injections followed by one injection every 2 months. The results of this study suggest that switching to ranibizumab for patients who do not achieve a dry retina while on aflibercept therapy may be beneficial. The use of a fixed monthly regimen rather than PRN, even after switching anti-VEGFs, or a treat and extend regimen, could also be considered. Further studies are warranted to evaluate longer term anti-VEGF switching outcomes.
Study limitations
As only one patient was recruited to the primary treatment failure group, the findings of the SAFARI study largely represent patients who achieved a suboptimal treatment response with aflibercept; comparison across subgroups was not possible. Furthermore, it is difficult to discern if the effect of the switch was solely due to the drug or also the posology. Due to the relatively small number of patients included, the findings of the secondary efficacy and exploratory assessments should be interpreted with caution.
Main conclusion
The SAFARI study demonstrated that switching to ranibizumab following an inadequate response or loss of efficacy with aflibercept led to a significant improvement in the primary efficacy endpoint of CSRT with ~60% achieving stabilised or improved VA. No new safety signals were identified. Therefore, patients with nAMD who have shown a suboptimal response to aflibercept may benefit from switching to ranibizumab.
Acknowledgments
This study and all costs associated with the development of this manuscript, including the article processing charges and open access fees were funded by Novartis Pharmaceuticals UK Limited. All named authors meet the International Committee of Medical Journal Editors criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole and have provided final approval for this version to be published. The authors acknowledge Arianna Psichas, PhD, and Danielle Sheard, MA (Cantab), Costello Medical, Cambridge, UK, for medical writing and editorial assistance in preparing this manuscript for publication, based on the authors’ input and direction, with funding from Novartis Pharmaceuticals UK Limited. The authors also thank the SAFARI study investigators for their help with conducting the study (from the UK: Konstantinos Balaskas, Ben Burton, Susan Downes, Clive Edelsten, Haralabos (Babis) Eleftheriadis, Richard Peter Gale, Sheena George, Faruque Ghanchi, David Gilmore, Robin Hamilton, Andrew Lotery, Geeta Menon, Nishal Patel, Ian Pearce, Sharmila Poovali, Priya Prakash, Cynthia Santiago, Simon Taylor, Saju Thomas, Mohan Varikkara, Deepali Varma, Meena Virdi, Gavin Walters, Michael Williams, Saad Younis, Rosina Zakri; from Germany: Nicole Eter, Frank Holz, Antonia Joussen, Wolfgang Lieb, Daniel Pauleikhoff, Barbara Schmidt, Walter Sekundo, Peter Wiedemann, Armin Wolf).
References
Footnotes
Contributors Substantial contributions to study conception and design as members of the study steering committee: RPG, IP, NE, FG, FGH, SS-V; substantial contributions to analysis and interpretation of the data: RPG, IP, NE, FG, FGH, SS-V, KB, BJLB, SD, HE, SG, DFG, RH, AL, NP, PP, CS, ST, DV, GW, MAW, AW, RZ, FI, FA; drafting the article or revising it critically for important intellectual content: RPG, IP, NE, FG, FGH, SS-V, KB, BJLB, SD, HE, SG, DFG, RH, AL, NP, PP, CS, ST, DV, GW, MAW, AW, RZ, FI, FA; final approval of the version of the article to be published: RPG, IP, NE, FG, FGH, SS-V, KB, BJLB, SD, HE, SG, DFG, RH, AL, NP, PP, CS, ST, DV, GW, MAW, AW, RHZ, FI, FA.
Funding Funding for this study was provided by Novartis Pharmaceuticals UK Limited.
Competing interests RPG: consultancy, educational grants and institutional research grants for Novartis and Bayer; IP: speakers’ fees, consulting fees and travel support from Novartis and Bayer; NE: grant/research support from Novartis and Bayer; speakers’ honoraria from: Novartis, Bayer, Allergan and Heidelberg Engineering; Advisory board member for: Novartis, Bayer, Allergan, Bausch and Lomb, Alimera Sciences and Roche; consultancy fees from Bayer; FG: consultant for Novartis and Bayer; advisory board member for Novartis and Bayer; speakers’ fees and research support from Bayer; FGH: financial support from: Acucela, Allergan, Bayer, Bioeq, CenterVue, Hoffmann-La Roche/Genentech, Heidelberg Engineering, Novartis, and Zeiss; Consultant for Acucela, Alcon, Bayer, Boehringer-Ingelheim, Hoffmann-La Roche/Genentech, Grayburg Vision, Heidelberg Engineering, Lin Bioscience, Novartis, Stealth Biotherapeutics, and Zeiss; SS-V: financial support from: Acucela, Alcon/Novartis, Allergan, Bayer, Bioeq/Formycon, Carl Zeiss MedicTec, CenterVue, Hoffman-La Roche/Genentech, Heidelberg Engineering, Katairo and Optos; consultant for: Alcon/Novartis, Bioeq/Formycon and Galimedix Therapeutics; recipient of gifts, honoraria, travel reimbursement, patent royalties and/or any other financial compensation from: Alcon/Novartis, Allergan, Bayer, Carl Zeiss MedicTec and Hoffmann-La Roche/Genentech; KB: travel grants and speakers’ fees from: Novartis, Bayer, Alimera, Topcon and Heidelberg Engineering; research grants from Novartis and Bayer; BJLB: advisory board and international conference attendance sponsored by Novartis and Bayer; SD: principal investigator on trials sponsored by: Novartis, Bayer, Roche, and Ely Lilly; speakers’ honoraria from: Novartis, Bayer, Roche and/or Eli Lilly; conference and travel support from Novartis and Eli Lilly; consultant for Hakko Kyowa and Circadian Therapeutics; grant/research support from Novartis; HE: travel grants and speakers’ fees from Novartis; advisory board member for Novartis; SG: speakers’ fees from Hoffman-La Roche and Novartis; advisory board member for, and travel grants from Bayer; DFG: travel grants from Novartis and Bayer; speakers’ fees from Novartis; RH: research support and travel grants from, and advisory board member for Novartis, Bayer, Allergan, and Ellex; AL: travel support to attend educational meetings from Bayer; NP: grant support and travel grants from Novartis and Bayer; PP: none declared; CS: speakers’ fees, consulting fees, conference and travel support from Novartis and Bayer; ST: travel grant and conference registration from Bayer; DV: travel grants from Novartis, Bayer and Allergan; Advisory board member for: Allergan, Novartis, Bayer, Alcon and Polyphotonix; investigator on clinical trials sponsored or funded by: Novartis, Bayer, Hoffmann-La Roche and Allergan; Speakers’ fees from Bayer and Novartis; GW: advisory board member for, and support to attend conferences from Novartis and Bayer; MAW: travel grants from Novartis, Bayer and Allergan; recipient of grant to develop medical education material on ophthalmology; AW: consulting fees from Novartis and Bayer; speakers’ fees from Novartis; advisory board member for Novartis; travel grants from Bayer; research support from Novartis and Bayer; RHZ: none declared; FI: employee of Novartis Pharmaceuticals UK Ltd; FA: employee of Novartis Pharmaceuticals UK Ltd.
Patient consent for publication Not required
Ethics approval This study was conducted in accordance with the current version of the applicable regulatory and International Conference on Harmonisation-Good Clinical Practice requirements, the ethical principles that have their origin in the principles of the Declaration of Helsinki, and the local laws of the countries involved.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Analysed data has been publically disclosed at clintrials.gov study number NCT02161575 and on the Novartis clinical trials portal at: https://www.novctrd.com/CtrdWeb/home.nov. For more details on the Novartis Position on Data Sharing and the eligibility criteria/process for patient-level data and clinical documents please go to www.clinicalstudydatarequest.com.
Linked Articles
- At a glance