Article Text

Abstract
Background/aims Proton beam radiotherapy and plaque brachytherapy are commonly applied in primary uveal melanoma (UM); however, their effect on chromosome 3 classification of UM by microsatellite analysis (MSA) for prognostication purposes is unknown, where the tumour is sampled post-irradiation. This study examined the prognostic accuracy of genotyping UM biopsied before or after administration of radiotherapy, by MSA.
Methods 407 UM patients treated at the Liverpool Ocular Oncology Centre between January 2011 to December 2017, were genotyped for chromosome 3 by MSA; 172 and 176 primary UM were sampled prior to and post irradiation, respectively.
Results Genotyping by MSA was successful in 396/407 (97%) of UM samples (196 males, 211 females; median age of 61 years (range 12 to 93) at primary treatment). There was no demonstrable association between a failure of MSA to produce a chromosome 3 classification and whether radiation was performed pre-biopsy or post-biopsy with an OR of 0.96 (95% CI 0.30 to 3.00, p=0.94). There was no evidence of association (measured as HRs) between risk of metastatic death and sampling of a primary UM before administration of radiotherapy (HR 1.1 (0.49 to 2.50), p=0.81). Monosomy 3 (HR 12.0 (4.1 to 35.0), p<0.001) was significantly associated with increased risk of metastatic death.
Conclusions and relevance This study revealed that successful genotyping of UM using MSA is possible, irrespective of irradiation status. Moreover, we found no evidence that biopsy prior to radiotherapy increases metastatic mortality.
- genetics
- neoplasia
- iris
- eye (globe)
- diagnostic tests/investigation
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Introduction
In the management of primary uveal melanoma (UM), patients are increasingly being offered prognostic biopsy for genetic analysis of their UM to estimate metastatic risk. Because of the low DNA yield from some small tumour samples, microsatellite analysis (MSA) is often used to determine the copy number variation (CNV) of chromosome 3 (Chr3) for prognostic purposes. MSA was first successfully carried out in UM by Tschentscher et al in 2000,1 and several studies have since confirmed its efficacy in accurately stratifying patients as having high or low metastatic risk.2–4
Most patients with uveal melanoma are treated with some form of radiotherapy.5–7 The most common radiotherapy modalities employed in UM are plaque brachytherapy (PRXT) and proton beam radiotherapy (PBR).7–10 Brachytherapy is most effective when used to treat small-to-medium sized UM where the thickness is ≤7 mm,11 whereas PBR can be used to treat UM that are larger or closer to the optic disc and the fovea, taking advantage of the Bragg peak and utilising a modified beam structure.12 Newer techniques, such as stereotactic radiosurgery with CyberKnife or Gamma Knife, achieve similar local control rates with eye retention to PBR but have a poorer visual prognosis post-treatment.13
The purpose of this study was to: (a) examine the mortality of patients with UM where Chr3 CNV was determined by MSA, (b) assess the effect, if any, of radiotherapy on successful Chr3 tumour classification and (c) establish whether sampling tumours before the administration of radiotherapy affects survival. Herein, we report MSA data from UM genotyped between 2011 and 2017 and including the largest cohort to date of UM samples obtained after PBR and PRXT, correlating these findings with genetic, histopathological and clinical data in addition to mortality.
Materials and methods
Tumour samples
A database query was carried out to identify all UM patients who were examined and treated at the Liverpool Ocular Oncology Centre (LOOC), Royal Liverpool and Broadgreen University Hospital Trust between January 2011 and December 2017, and who had genetic testing of their UM performed by MSA. All patients underwent a full systemic and ophthalmic examination at the LOOC and clinical, histopathological, genetic and follow-up data were collected. PBR was administered at the Clatterbridge Cancer Centre at a dose of 56 Gy over four consecutive days. Ruthenium PRXT delivered an apical tumour dose of a minimum of 80 to 90 Gy. Trans-retinal and trans-scleral tumour biopsy samples were obtained using methods described previously14 either before or after radiotherapy. A single histology cytospin was produced for each biopsy and stained with May-Grünwald-Giemsa, as previously described.15 Confirmation of the presence of UM cells in the biopsy sample was undertaken by an experienced ophthalmic pathologist (SEC). All surgical resection samples (eg, enucleation, local resection, endoresection and iridocyclectomy) were processed using methods described previously.16–19 Peripheral blood samples were collected at the time of surgical procedures to provide constitutional DNA used as a control in the MSA analysis.
This study was conducted in accordance with the tenets of the Declaration of Helsinki and Good Clinical Practice Guidelines. All samples and data were provided by the Ocular Oncology Biobank (REC Ref 16/NW/0380). All patients had provided informed consent for the use of their samples and data in research.
DNA extraction and genotyping by microsatellite analysis
DNA extraction was performed on UM tissue and blood samples as described by Lake et al (modified QIAamp and DNeasy Blood and Tissue kit; Qiagen, Crawley, UK).20 DNA was resuspended in TE buffer (10 mM Tris-HCl pH 8.0, 0.1 mM EDTA) (Life Technologies Ltd, Carlsbad, California) and quantified using fluorometric methods (Invitrogen Qubit fluorometer and broad-range DNA quantification assay; Life Technologies, Carlsbad, California; Glasgow, UK). All samples tested by MSA had a DNA yield between 2 to 20 ng/µl. MSA was performed using a modified protocol from Thomas et al.4 Briefly, genetic analysis was carried out using a polymerase chain reaction (PCR)-based technique assessing eight polymorphic microsatellite markers on Chr3; four microsatellite loci on 3p and four microsatellite loci on 3q (online supplementary table 2). All PCR steps were carried out on a G-Storm GS1 Thermal Cycler (Genetic Research Instrumentation Ltd, Essex) using the following conditions: initial activation step at 95°C for 15 min; and then 35 cycles of 94°C for 30 s, 56°C for 90 s and 72°C for 60 s with a final extension for 30 min at 60°C, cooling at 10°C. PCR products were loaded in 8.5 µl Hi-Di Formamide containing 1 µl LIZ500 size standard analysed using the ABI 3500 Genetic Analyzer. Fragment analysis was completed using GeneMapper V.3.5 software (Applied Biosystems). An allelic ratio (AR) was calculated by normalising the allele peak area of the tumour against the corresponding blood sample (the control sample). The genotype of a locus was assigned based on the calculated AR: AR ≥2.5, loss of heterozygosity (LOH); AR 2.49 to 1.4, allelic imbalance (AI) or AR ≤1.39, no loss of heterozygosity (NLOH).
Supplemental material
Chr3 classification
A UM was classified as monosomy 3 (M3) when two or more microsatellites on chromosome arm 3p and two or more microsatellites on chromosome arm 3q showed LOH. UM were classified as disomy 3 (D3) when two or more microsatellites on chromosome arm 3p and two or more microsatellites on chromosome arm 3q showed NLOH. Partial loss (PL) of Chr3 was assigned if at least two markers were lost on one chromosome arm, when the other arm showed NLOH. UM, in which two or more microsatellites on chromosome arm 3p and two or more microsatellites on chromosome arm 3q showed AI, were classified as AI. When UM had two NLOH and two LOH on each arm, a tumour was classified as M3. UM were considered as ‘unclassifiable’, if none of the above conditions could be met15 (online supplementary figure 1).
Supplemental material
Statistical analyses
Follow-up (years) was calculated from the date of the first diagnosis of the primary UM until death or study closure on 17th January 2019. Statistical analyses were carried out using IBM SPSS Statistics V.24 (https://www.ibm.com/products/spss-statistics), Microsoft R 3.5.1, and the packages rms, cmprsk and mstate (https://mran.microsoft.com/). Because of the small sample size, to control the false discovery rate, the statistical significance was defined as p<0.001 (two-sided).
Results
Patient demographics
From January 2011 to December 2017, 407 UM patients receiving treatment at the LOOC underwent genotyping for Chr3 using MSA (online supplementary table 1). The study cohort comprised 196 males and 211 females with a median age of 61 years at primary management (range 12 to 93 years). The median follow-up was 54 months (range 5 to 368 months). The UM had a median largest basal diameter of 11.2 mm (range 1.8 to 20.8 mm) and a median ultrasound height of 3.1 mm (range 0.9 to 18.5 mm). UM involving the ciliary body was 53/407 (13%) and 5/407 (1%) had extraocular extension. The tumour size category according to the eighth AJCC (American Joint Committee on Cancer) TNM classification system was: T1 in 186 cases (46%), T2 in 146 cases (36%), T3 in 59 cases (14%) and T4 in 16 cases (4%). On histological examination, 116/407 (29%) UM samples contained epithelioid cells.
Supplemental material
At the time of study closure on 17th January 2019, 347/407 (85%) patients were alive, 35/407 (9%) patients had developed or died from metastatic disease, 20/407 (5%) patients died from other or unknown causes and 5/407 (1%) patients were lost to follow-up.
Nine of the 407 UM samples (2%) analysed were diagnostic biopsies: 6/9 went on to have secondary enucleations; 1/9 had a subsequent endoresection with 2/9 receiving no further treatment.
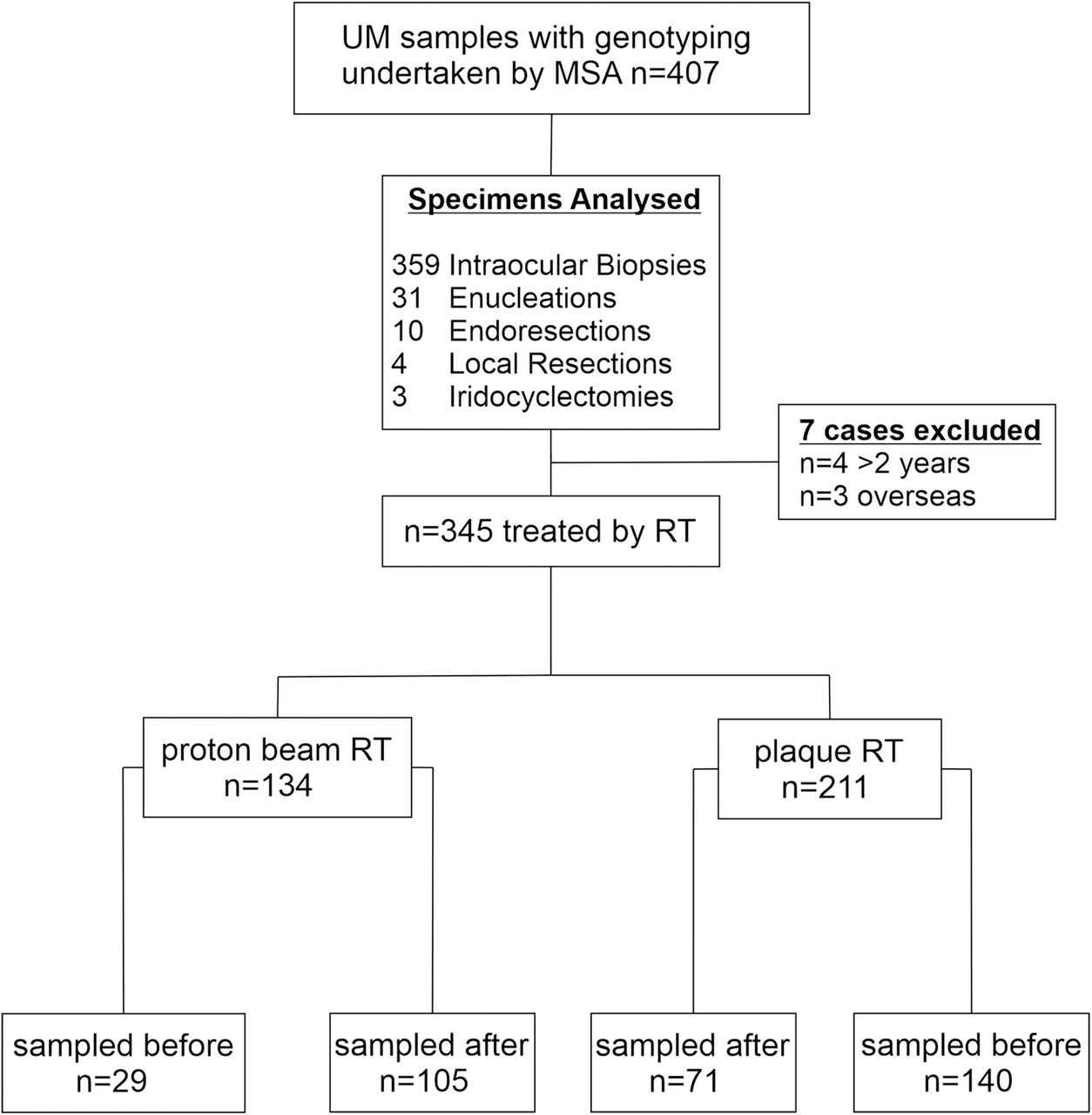
The 407 examined UM samples consisted of: 359 intraocular biopsies, 31 enucleations, 4 local resections, 10 endoresections and 3 iridocyclectomies (figure 1). Seven cases analysed were excluded from the time to biopsy analysis following radiotherapy due to sampling more than 2 years after radiotherapy treatment (n=4) and overseas patients who were lost to follow-up (n=3), leaving 345 cases that had received radiotherapy.

Flowchart of specimens examined in the present study; n=407 samples examined, n=353 received radiotherapy either n=136 proton beam radiotherapy or n=217 ruthenium plaque radiotherapy; n=174 samples were taken before administration of either proton beam or plaque radiotherapy of which n=6 failed genotyping; n=179 samples were taken after administration of either proton beam or plaque radiotherapy of which n=6 failed genotyping. MSA,microsatellite analysis; RT, radiotherapy; UM, uveal melanoma.
Of the 345 UM patients that received radiotherapy, 169 (49%) and 176 (51%) were sampled pre-radiotherapy and post-radiotherapy, respectively. The median time interval between biopsy and PBR was 32 days with the range spanning from 66 days preoperatively (including some diagnostic biopsies) to 284 days postoperatively. Similarly, the median time interval between biopsy and PRXT was 0 days with the range spanning from 49 days preoperatively to 129 days postoperatively. Of the 176 samples analysed post-radiotherapy only six (3%) failed to provide a Chr3 classification. The median time to biopsy of these six cases was 36.5 days (range 24 to 52)
Microsatellite analysis
UM samples successfully genotyped were 395/407 (97%) according to the MSA classifications described in Materials and Methods: 97 (24%) UM were M3 (20% of AJCC 1, 48% AJCC 2, 20% AJCC 3 and 9% AJCC 4), 256 (63%) UM were D3 (53% AJCC 1, 32% AJCC 2, 13% AJCC 3 and 2% AJCC 4), 16 (4%) UM were PL, all loss of 3q, (50% AJCC 1, 31% AJCC 2 and 19% AJCC 3) and 26 (6%) UM were AI (54% AJCC 1, 35% AJCC 2, 8% AJCC 3 and 3% AJCC 4). Further, 12 (3%) UM were considered ‘unclassifiable’ (67% AJCC 1, 25% AJCC 2 and 8% AJCC 3). Of these, 6/12 (50%) were sampled pre-radiotherapy and 6/12 (50%) post-radiotherapy. Of the 35 UM patients who died of metastatic UM, 27 (77%) had M3 UM, 2 (6%) had loss of 3q, 5 (14%) were D3 and 1 (3%) was ‘unclassifiable’.
For samples taken pre-radiotherapy and post-radiotherapy, the genetic results were comparable and there was no significant difference in the number of UM cases for which Chr3 data (either M3, D3, PL or AI) was obtained (X2 p=0.099).
Cox analysis
The data set comprised of 345 complete observations. The variables used in the analysis are shown in table 1.
Characteristics of 345 UM cases genotyped either before or after RXT
A Cox proportional hazards model21 was fitted to assess the impact on metastatic death hazard rate of biopsy sampling before or after radiotherapy and Chr3 classification. Contrasts were specified so D3 was the baseline level, and the hazard rate for the other four Chr3 classes are specified in relation to D3.
The hypothesis of the proportionality of hazards was assessed.21 Table 2 shows the HRs, p values of the z statistics and proportionality of hazards test for each factor. There is evidence only of a difference between hazard rates associated to M3 and D3.
Cox model statistics
Because of the small number of metastatic deaths, the 95% CIs on the HRs of Chr3 classes were wide; in the case of AI, no metastatic events occurred, so the model parameters could not be identified.
The C-index21 with 95% CI was 0.78 (0.69 to 0.87), denoting good discrimination performance.
Logistic regression
A logistic regression model21 was fitted to assess the impact of biopsy sampling before or after radiotherapy on the success of genotyping. There was no evidence of this factor affecting the success of genotyping by MSA, with OR of 0.96 (95% CI 0.30 to 3.0, p=0.94)
Cumulative incidence analysis
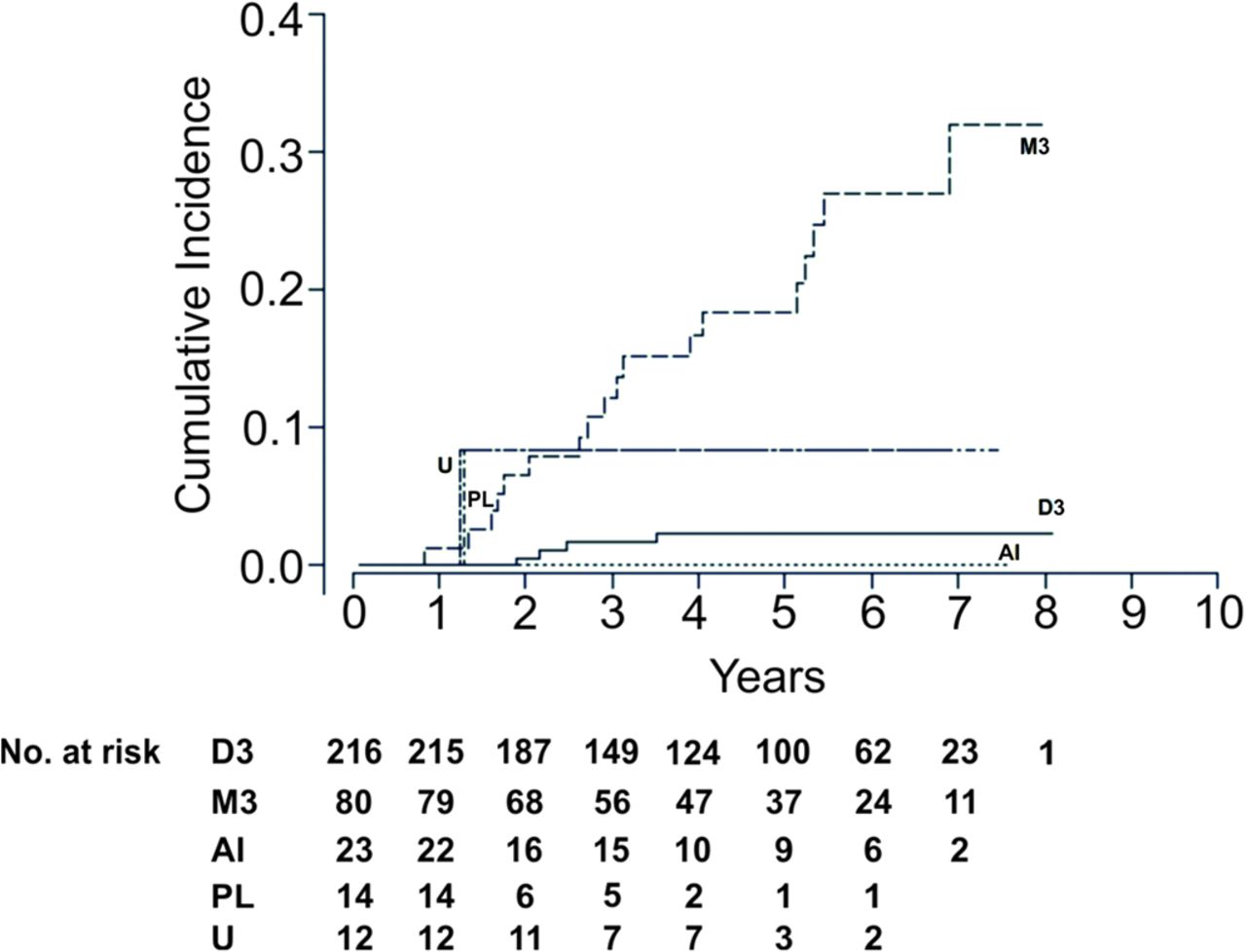
Figure 2 shows the cumulative incidence (c.i.) of metastatic death by Chr3 levels. No metastatic death events were observed in the AI group, so the cumulative incidence was zero. It should be noted that the Loss 3q c.i. and Unclassifiable c.i. were essentially the same, overlapping with M3 c.i. up to about 3 years. Furthermore, the estimated 95% confidence limits on Loss 3q c.i. and Unclassifiable c.i. (not shown to avoid clutter) were so large that they enclosed both the M3 and D3 c.i.

{kind=link}
{kind=link}
Cumulative incidence functions and number at risk for each time interval, by Chr3 level. AI, allelic imbalance; Chr3, chromosome 3; D3, disomy; M3, monosomy; PL, partial loss 3q; U, unclassifiable.
Gray’s K-sample test statistic22 for comparing the c.i. across the Chr3 levels is G=34 (p<0.001). This result qualitatively agreed with the Cox analysis, and the difference could be attributed mostly to M3 and D3.
Discussion
To our knowledge, this is the largest series of post-radiotherapy UM samples genotyped for Chr3 status to date. We have shown that MSA can be used to establish Chr3 status in 97% of cases. Taking a biopsy before administration of radiotherapy did not increase the risk of metastatic death. Furthermore, neither PBR nor PRXT affected genotyping classification.
MSA is a molecular technique that can accurately determine Chr3 status in small UM biopsy samples with low DNA concentrations.23 In our study, genotyping was successful in 97% of all cases examined resulting in classification into two main groups, M3 and D3. The Chr3 classifications show evidence of an increased (with respect to D3) hazard of metastatic death in the case of M3, but no evidence for the other classes. Effects could not be reliably estimated in the case of AI because of a lack of associated metastatic death events. In all cases, the small number of events translates into large uncertainties in the estimation of HRs. Analysis of cumulative incidences provides qualitatively similar results. Of interest, a large proportion of M3 classifications by MSA were observed in AJCC stages 1 and 2 highlighting that smaller tumours are not immune from being at high risk of developing metastases. This is consistent with the findings of Damato et al who demonstrated many small tumours showed risk factors for metastasis, such as M3.24 Cases with PL of Chr3 and AI were also observed. PL of Chr3 in this study occurred exclusively as loss of 3q in 4% of the UM genotyped. Thomas et al also reported PL of Chr3 in 4% of UM cases analysed by MSA, while in the study by Shields et al PL of Chr3 was detected in 27% of UM cases.4 23 The incidence of PL of Chr3 varies greatly in the literature, with some studies reporting it to be between 0% and 48% by MSA and other techniques.3 It is suggested that PL of Chr3 is caused by tumour heterogeneity. Cytomorphological heterogeneity is well documented in posterior UM and has led to concerns that extracting a biopsy from a single site may not be representative of the whole tumour.25 25 Heterogeneity of gene loci dosage quotients was reported in a study utilising multiplex ligation-dependent probe amplification (MLPA) to examine CNV in UM; however, this did not affect the overall CNV classification.26 In the present study, eight UM patients underwent a subsequent enucleation following biopsy, and the prognostic results were concordant for each of these. Similarly, Coupland et al reported concordant Chr3 data for patient-matched samples in 28 UM cases that were initially biopsied and subsequently resected.27
In this study, no patients with AI developed metastatic disease; however, because of the small number of cases with this classification, no conclusive association of AI with mortality could be made. AI was first reported by Tschentscher et al who consistently observed allele ratios that fell just below the cut-off thresholds for gain or loss.1 They reasoned that this may be the result of clonal heterogeneity or more focal dosage changes. Thomas et al demonstrated that UMs with AI showed survival similar to those with M3 UM, and thus were associated with a poor prognosis.4 In their study, UM were defined as ‘AI’ if at least two markers showed AI even if the remaining markers were LOH, which in the current study would have been classified as M3.
The impact of taking a biopsy before or after radiotherapy on the success of genotyping was also assessed, using a logistic regression model. The OR shows no evidence of an effect, but it should be considered the small number of failed genotyping entries, which tends to bias the classification towards the class with the largest number of entries (in this case, successful genotyping). These data are, however, consistent with the results of Hussain et al who demonstrated that genetic analysis of UM by MLPA and MSA after treatment with PBR provided Chr3 classifications predictive of metastasis free survival.14 Similarly, in a study by Coupland et al four UM samples obtained both pre-radiotherapy and post-radiotherapy showed concordant genetic results, demonstrating successful genotyping of irradiated specimens.27 Another analysis by Wackernagel et al utilised array comparative genomic hybridisation to test samples pre-radiotherapy and post-radiotherapy; five patients had genetic analysis done before and after radiotherapy, and their results were also completely concordant, thus confirming the suitability of these samples for genotype analysis.28 This was not the case in the study by Dogrusöz et al who used karyotyping and fluorescence in situ hybridisation to genotype irradiated tumours.29 Analysis of these samples was largely unsuccessful mainly due to tumour shrinkage and necrosis associated with irradiation, and in comparison to other studies, there was a significantly longer time from irradiation until enucleation (5 to 146 months). Most recently, Matet et al demonstrated genetic concordance of clinically relevant chromosomes in 94% of matched biopsies taken before PBR and a subsequent endoresection taken less than 3 months following radiotherapy.30
To our knowledge, this is the first study of its kind to examine whether taking a biopsy before treatment by radiotherapy is associated with death from metastatic UM. An ex vivo study performed by Glasgow et al demonstrated iatrogenic dissemination of tumour cells following transvitreal biopsy.31 There have also been other case reports and series of suspected dissemination, which has contributed to the reluctance of some ophthalmologists to take diagnostic and prognostic biopsies.32–35 In this study, Cox analysis shows no evidence that a biopsy taken before or after radiotherapy affects the metastatic event hazard rate. This is consistent with the findings of a recent study by Bagger et al where a retrospective nationwide audit of 1637 UM patients demonstrated that melanoma-specific mortality was not increased in biopsied patients as compared with non-biopsied patients.36
One of the limitations of this study was the relatively short follow-up for some of the cases included in analyses, with 5 months being the shortest interval; however, it was still possible to show statistically significant differences between genotype results.
Although, MSA is a highly successful genotyping technique at our centre for UM samples yielding a small amount of DNA, next-generation sequencing (NGS) panels are increasingly being used for this type of analysis. This comprises broader pan cancer panels,37 38 whole exome sequencing39 and targeted NGS panels,40 including a bespoke NGS UM panel quite advanced in its development at our own centre,41 based on The Cancer Genome Atlas mutational data,42 which also successfully obtains reliable genotyping data in previously irradiated tumours.
In summary, we have shown that MSA is a reliable genotyping technique that can provide Chr3 classification in irradiated and non-irradiated UM. In addition, melanoma-specific mortality is not increased when UM are biopsied prior to radiotherapy.
Acknowledgments
The authors would like to thank and acknowledge the Biomedical Scientists of the Ophthalmic Pathology team at the Liverpool Clinical Laboratories, Mr Simon Biddolph and Mrs Anna Ikin and Mr Gary Cheetham for maintaining of the database of the Liverpool Ocular Oncology Centre based at the Liverpool University Hospitals, UK.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors ST, SEC, HK: Design of study; ST: Undertaking microsatellite analysis; ST, SEC, AT, AE, HK: Evaluation of laboratory data with clinical data and outcomes; All authors: Interpretation of results; All authors: Manuscript writing; All authors: Critical review of manuscript.
Funding This work was supported by the Eye Tumour Research Fund Charitable Funds, Royal Liverpool University Hospital, UK, grant number (A091/CF).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Approval for the study was obtained from the Health Research Authority South Central - Hampshire B Research Ethics Committee (REC ref 15/SC/0611).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Deidentified participant data is available from the corresponding author upon reasonable request. ORCID ID. 0000-0001-7693-7279, or sophie.thornton@liv.ac.uk. Conditions of reuse dependent upon ethical approval and appropriate data transfer agreement.