Article Text

Abstract
Cataract is the most common cause of blindness in the world; during infancy and early childhood, it frequently results in visual impairment. Congenital cataracts are phenotypically and genotypically heterogeneous and can occur in isolation or in association with other systemic disorders. Significant progress has been made in identifying the molecular genetic basis of cataract; 115 genes to date have been found to be associated with syndromic and non-syndromic cataract and 38 disease-causing genes have been identified to date to be associated with isolated cataract. In this review, we briefly discuss lens development and cataractogenesis, detail the variable cataract phenotypes and molecular mechanisms, including genotype–phenotype correlations, and explore future novel therapeutic avenues including cellular therapies and pharmacological treatments.
- embryology and development
- genetics
- lens and zonules
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Introduction
Cataract is the most common, but treatable cause of blindness in the world. Recently, during the 70th World Health Assembly (October 2019), WHO estimated that 2.2 billion people are visually impaired around the world, out of which 65.2 million people are affected with cataract (https://www.who.int/publications-detail/world-report-on-vision). Congenital cataracts are detected at birth or during the first decade of life. WHO estimated that >14 million children are bilaterally blind from cataract, representing >50% of all causes of blindness globally.1 Congenital cataracts are present in 1–6/10 000 live-births in developed countries and 5–15/10 000 births in developing countries. They are a prominent cause of vision loss in infants and children.2 3 Most vision loss is due to amblyopia, but some are due to postoperative complications such as glaucoma and retinal detachment.
Approximately half of the congenital cataracts are characterised as inherited and are a clinical feature of nearly 200 syndromic genetic diseases,4 including for instance diabetes and cholesterol metabolism diseases. Congenital cataract was the first autosomal disease to be genetically mapped in humans5 and has subsequently been shown to be associated with considerable genetic and phenotypic heterogeneity.6–8 Several distinct phenotypes have been identified in families with autosomal-dominant congenital cataract based mainly on the location and appearance of the opacification in the lens: anterior polar, posterior polar, nuclear, lamellar, coralliform, blue dot (cerulean), cortical, pulverulent and polymorphic.9–11 The majority of the inherited cataracts are autosomal dominant with complete penetrance, but variable expression, autosomal recessive and X-linked inheritance patterns are less frequent.
Over the last 10 years, enormous progress has been made in elucidating the molecular basis of congenital cataract, with causative mutations identified in genes encoding many different proteins including intracellular lens proteins (crystallins), membrane gap junction proteins (connexins), water channel proteins (aquaporins), cytoskeletal proteins (eg, BFSP1 (filensin), BFSP2 (phakinin) and vimentin) and transcription factors (TFs) (eg, FOXE3, PAX6, PITX3 and MAFA) (table 1). Recent advances in molecular genetics, particularly next-generation sequencing, has improved molecular diagnosis in the clinic.12
Genes implicated in inherited cataract
Lens embryology and morphology
The ocular lens is a unique model to understand the important aspects of embryonic development, signalling, induction, cell differentiation, cell physiology, biochemistry, organelle degradation and cellular longevity. Several reviews can provide a detailed description.6 13 14
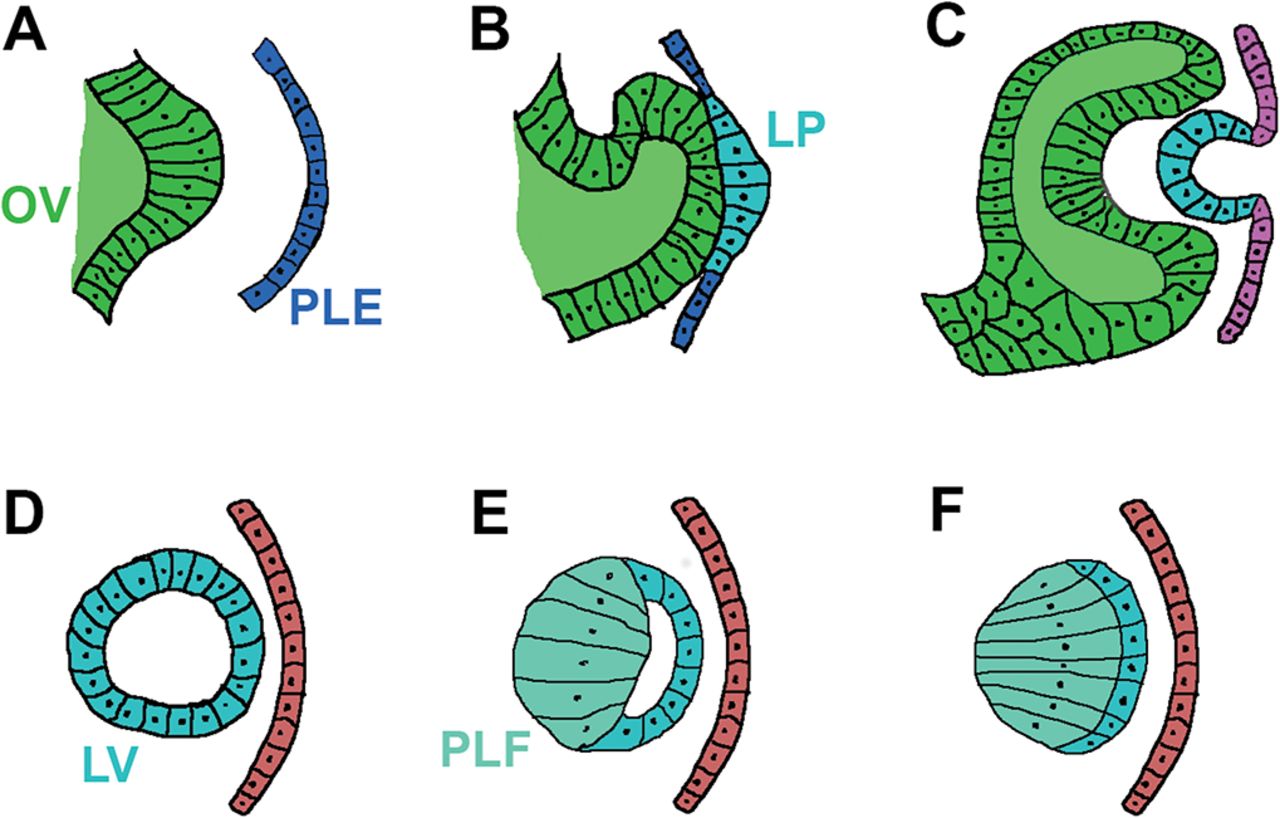
During gastrulation (day 22, Carnegie stage 9 in human development), a single eye field forms in the middle of the anterior neural plate, which separates into two optic vesicles and further induce the nearby surface ectoderm to form the lens placode (a precursor of the lens) by day 28 (Carnegie stages 12–13) (figure 1A,B). At this stage, a series of inductive interactions begin to shape the eye, orchestrated by signalling molecules such as bone morphogenetic proteins and fibroblastic growth factor 2, as well as TFs such as OTX2, PAX6 and PITX3.15 This programme can be mimicked in culture conditions to determine the differentiation pathway of pluripotent cells and their derivatives.16

Schematic diagram of the developing lens. (A) The OV approaches the PLE at human age 4 weeks, embryo length 3–5 mm, carnegie stage 12. (B) Contact between OV and PLE results in the LP formation at human age 4–5 weeks, embryo length 4–6 mm, carnegie stage 13. (C) LP invagination produces a lens pit at Carnegie stage 14, week 5, 31–35 days. (D) Lens pit detaches from the surface ectoderm and the LV is formed at human age 5–6 weeks, 7–9 mm embryo length, carnegie stage 15, day 35–38. (E) Posterior lens vesicular cells elongate toward the anterior epithelial cells to form PLFs. (F) The embryonal nucleus is formed following the obliteration of the LV lumen at human age 6 weeks, 8–11 mm embryo length, carnegie stage 16. LP, lens placode; LV, lens vesicle; OV, optic vesicle; PLE, presumptive lens ectoderm; PLF, primary lens fibre.
The lens placode invaginates to form the lens pit (figure 1C), which makes a complete circle of cells and detaches from the surface ectoderm to develop into the lens vesicle (figure 1D). By the end of week 4 (Carnegie stages 10–13), the cells from the posterior vesicle start elongating towards the anterior epithelial cell layer to become the primary lens fibres that fill the lens vesicle and later become the embryonic nucleus of the mature lens (figure 1E). The portion of the optic vesicle that faced the lens placode gives rise to the retina. The retina, in turn, provides inductive signals that regulate the growth and apical-posterior axis of the lens. In the early optic cup stage, the lens vesicle releases signals that induce the overlying surface ectoderm to differentiate into the corneal epithelium. Around weeks 6–7 (Carnegie stages 16-19), lens fibres start to develop from the epithelial cells located at the equator where they begin to elongate and differentiate into the secondary lens fibres (fetal nucleus) of the developing lens (figure 1F). Around week 8 (Carnegie stage 20), the Y-shaped suture appears at the anterior and posterior poles of the embryonic nucleus of the lens as a result of the terminal ends of the secondary lens fibres abutting each other. The newly differentiated fibre cells continue to grow throughout life. During this process of terminal differentiation, fibre cells remove their nucleus and other cell organelles to minimise light scattering.
Cataract phenotypes and modes of inheritance
The congenital cataract phenotype broadly reflects spatiotemporal insults experienced by the developing lens. Broadly these phenotypes can be divided into eight distinct clinical appearances: (1) nuclear (figure 2A), (2) anterior polar (figure 2B), (3) posterior polar (figure 2C), (4) lamellar, (5) coralliform, (6) blue dot (cerulean – figure 2D), (7) cortical (figure 2E) and (8) pulverulent (figure 2F).9–11
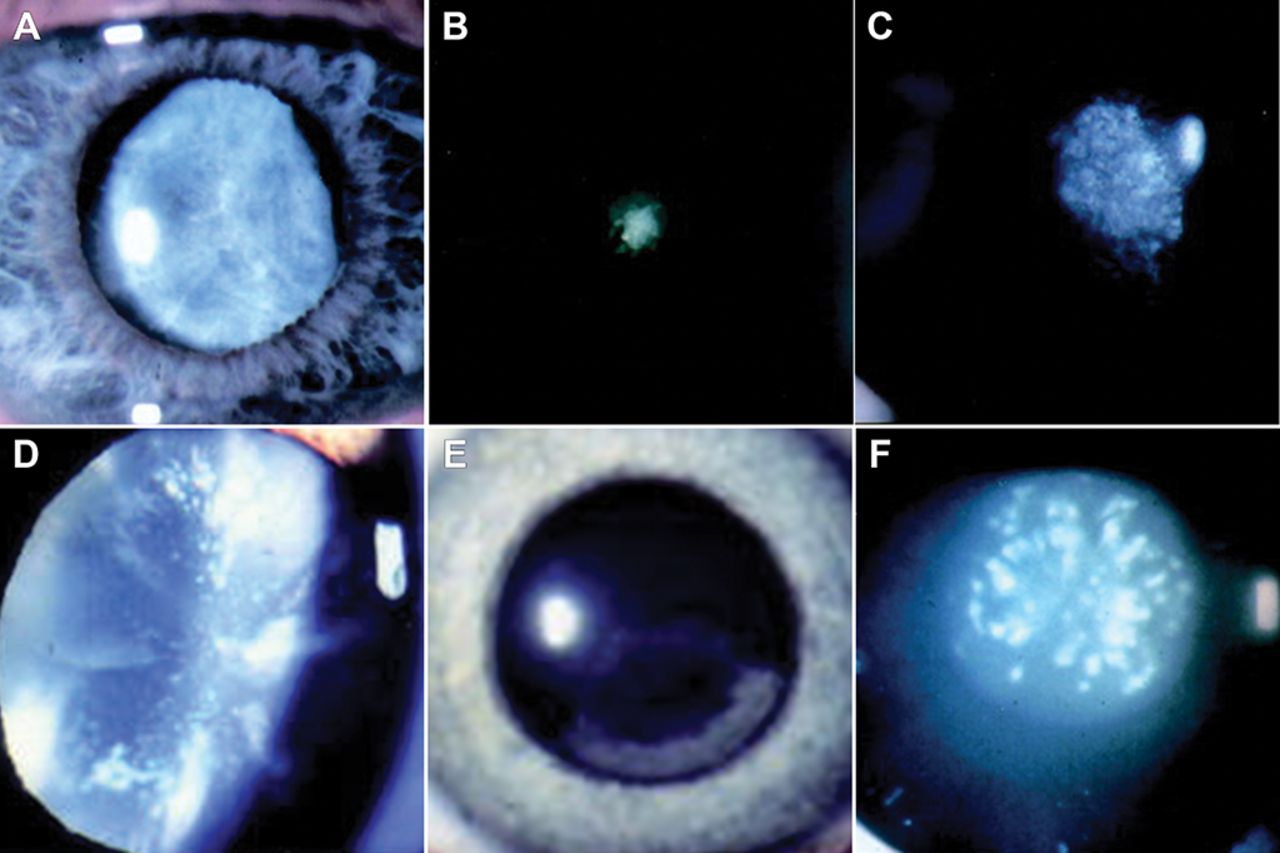
Slit-lamp presentation of examples of inherited cataract phenotypes. (A) Nuclear cataract, (B) anterior polar cataract, (C) posterior polar cataract, (D) blue dot cataract (Cerulean), (E) cortical cataract and (F) pulverulent cataract.
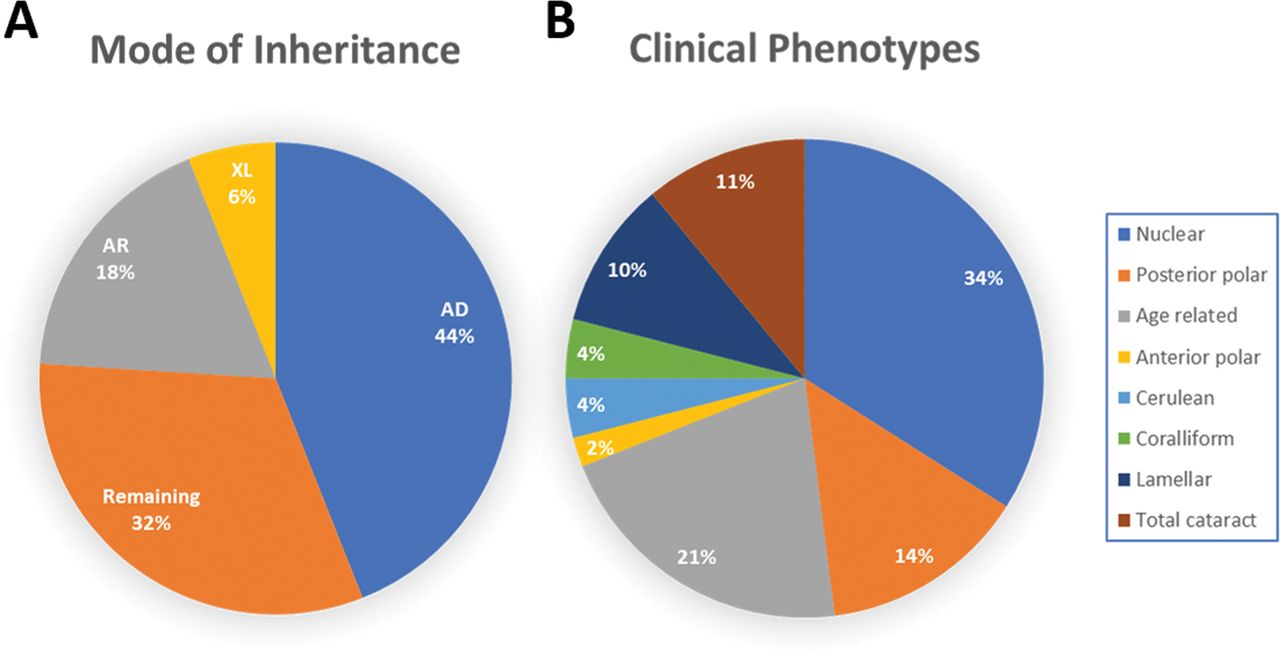
There is considerable phenotypic variation in autosomal cataract, although there have been few systematic studies recording differences for specific mutations.8 To date (August 2019), 1314 novel and recurrent disease-causing sequence variants have been identified (http://cat-map.wustl.edu/), with a well-defined distinct phenotype observed in 366 cases as shown in figure 3A. Interestingly, the most frequent phenotype was nuclear cataract (34%), followed by posterior polar (14%), total cataract (11%), lamellar (10%), cerulean and coralliform both (4%), and anterior polar (2%), and the remaining were age-related cataracts (21%) (figure 3B).

Frequency pie charts. (A) Frequency of various mode of inheritance. (B) Frequency of cataract phenotypes in families seen to date. AD, autosomal dominant; AR, autosomal recessive; XL, X-linked.
The most common mode of inheritance is autosomal dominant (44%), followed by autosomal recessive (18%), X-linked recessive (6%) with the remaining 32% being sporadic with no clear family history (https://cat-map.wustl.edu/) (figure 3A).17 18 Improvements in molecular diagnosis using next-generation sequencing will allow a better understanding of the mode of inheritance in sporadic cases, to establish whether the variant is homozygous/recessive or heterozygous/dominant.
Molecular genetics
Disease-causing sequence variants in 115 genes have been identified in isolated and syndromic inherited cataract. Thirty-eight genes are responsible for distinct isolated cataract and can be divided into five main groups based on the encoded proteins: (1) crystallins, (2) membrane proteins, (3) transcription factors, (4) cytoskeletal proteins and (5) gene products with special roles in the lens (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cataract-causing variants by gene. Total number of 1314 disease-causing variants (novel and recurrent) are shown in various highly expressed genes to date (http://cat-map.wustl.edu/), including crystallins, gap junction proteins, membrane proteins, developmental and cytoskeletal proteins in the lens; remaining 476 variants are found in various other genes important for the normal function of the crystalline lens.
Crystallins
Crystallins (α, β and γ) account for nearly 90% of all lens proteins; they are crucial for the lens optical properties and its function, for the remarkable resilience to post-translational modifications due to ageing19 and the environment to maintain lens transparency.20 21
Alpha-crystallins are members of the small heat shock protein family, which are molecular chaperones protecting lens proteins and enzymes from aggregation, which could otherwise lead to lens opacification.20 Alpha-crystallin comprises two related subunits (αA polypeptide and αB polypeptide) encoded by CRYAA on chromosome 21q22.3 and CRYAB on chromosome 11q22-q22.3, respectively.22 Variants in CRYAA cause both autosomal-dominant and autosomal-recessive cataracts and account for 25.8% of all crystallin mutations. CRYAA is primarily expressed in the lens; there is also extralenticular expression detected in mouse retina and cornea.23–25
CRYAB is expressed in the lens epithelial cells and in the retina, skeletal muscle, heart, kidney and brain.24 26 Mutations in CRYAB cause not only cataract but also myopathies there being heart and lens-specific enhancers to regulate its expression in these tissues. In 2001, Berry and colleagues found the first dominant heterozygous mutation in a large family with posterior polar cataract.27 To date, 22 mutations have been reported in the CRYAB gene in both autosomal-dominant and autosomal-recessive cataract. Heterozygous missense variants have also been described in patients with desmin-related myopathies and cardiomyopathy.28
The βγ-crystallins are derived by gene duplication and belong to a large superfamily that includes such proteins as AIM1.24 They consist of four homologous Greek key motifs organised into two domains. The β-crystallin family comprises four acidic (A) and three basic (B) forms encoded by genes—CRYBA2, CRYBA1 and CRYBB3, CRYBB2, CRYBB1 and CRYBA4, respectively. γ-Crystallin is encoded by the γ-gene cluster on chromosome 2q33–35 encompassing genes γA to γD. Fewer mutations are found in the γA (CRYGA) and γB (CRYAGB) compared with γC (CRYGC) and γD(CRYGD).29–32 Interestingly, most of the variants in CRYGC and CRYGD genes cause nuclear cataract and coralliform phenotypes inherited dominantly. A single γs-crystallin gene resides on the long arm of chromosome 3q25-qter; mutations in this gene are mainly causing autosomal-dominant disease with various phenotypes. Crystallins is the most prevalent proteins in the lens; nearly 294 variants have been found in crystallins, which account for nearly 22.4% of all the inherited cataract variants found to date (figure 4).
Membrane proteins
Connexins
A sophisticated cell-to-cell communication network is important in maintaining lens cell homeostasis. In the developing lens, this communication is maintained via gap junction channels which allow the flow of ions, second messengers and metabolites between lens fibre cells. In the lens, these channels are made up of three connexin isoforms: GJA1 (Cx43), GJA3 (Cx46) and GJA8 (Cx50). Six connexin molecules assemble to form a hemichannel or connexon. Each connexon is either made from a single type of connexin, or from more than one type, which leads to either homomeric or heteromeric hemichannels, respectively. These hemichannels dock with a counterpart in an adjacent cell to make a gap junction channel linked by their extracellular loops.33 34 GJA1 is expressed only in the lens epithelial cells during early stages of lens development, but not associated with lens pathology.35 36 Fifty-five heterozygous variants and 1 homozygous variant have been found in GJA3, with various associated lens phenotypes, including pulverulent, nuclear, lamellar, coralliform and total (http://cat-map.wustl.edu/). To date, 90 heterozygous variants have been described in families with autosomal-dominant cataract, and a single homozygous variant in autosomal-recessive cataract has been found in GJA8, associated not only with inherited cataract but also age-related cataract and other eye anomalies including microcornea, microphthalmia and corneal opacification.37
MIP
MIP26 is the major intrinsic protein of the lens, encoded by AQP0, a member of the ubiquitous family of water channel proteins called aquaporins that allow rapid movements of water across cell membranes. MIP is highly expressed in terminally differentiated lens fibre, comprising nearly half of the total lens fibre cell membrane proteins.38 Berry and colleagues39 40 identified two autosomal-dominant variants (G134E and T138R) leading to polymorphic and lamellar cataract. So far, 37 heterozygous variants have been found in MIP causing autosomal-dominant cataract.7
LIM2
LIM2 is a lens-specific integral membrane protein, also referred to as MP19. This gene encodes an eye lens-specific protein found at the junctions of lens fibre cells, where it may contribute to cell junctional organisation. It acts as a receptor for calmodulin and may play an important role in both lens development and cataractogenesis. Mutations in LIM2 have been associated recessive cataracts and age-related cataracts.41–43
Transcription factors
TFs including PAX6, FOXE3, HSF4, MAF and PITX3 among many others play an important role in lens development.13 14 PAX6, the paired-box gene is one of the key players in vertebrate eye development. In the lens, PAX6 plays a key role in the regulation of lens-specific crystallins.44 45 and thus is an important TF in lens development. Heterozygous null mutations in PAX6 typically give rise to aniridia often with associated lens opacities but missense changes may result in milder phenotypes including foveal hypoplasia, Peters anomaly and isolated cataract.46 47
FOXE3 is a forkhead-box TF, required for morphogenesis and differentiation of the anterior segment of the eye.48 Semina et al reported the first human variants in this gene causing anterior segment mesenchymal dysgenesis and congenital cataracts.49 More than 20 homozygous and heterozygous variants have been reported, displaying severe developmental eye anomalies including cataract.50
MAF, is a basic region leucine zipper (bZIP), an oncogene, which is expressed in early lens development.51 MAF regulates the expression of the lens crystallins.52 Mutations in MAF are not only responsible for cataract and ocular abnormalities but also cause Aymé-Gripp syndrome.53 54 Recently, a missense mutation in MAFA gene has been reported to cause autosomal-dominant insulinomatosis and diabetes mellitus along with cataract and glaucoma.55
PITX3 gene is a member of the REIG/PITX family of homeobox TFs.56 To date, 26 variants in PITX3 have been identified (including a hot spot in exon 4, c.640_656dup17bp) to cause mainly posterior cataracts and anterior segment dysgenesis in different ethnicities.57
Cytoskeletal proteins
The cytoskeleton of a cell comprises three major filaments: microfilaments, microtubules and intermediate filaments. In the lens, two beaded filaments, a type of intermediate filaments, BFSP1 (filensin) and BFSP2 (phakinin), are expressed.58 Several variants in BFSP2 lead to sutural opacities and nuclear cataract in association with BFSP1 variants. NHS (Nance-Horan Syndrome) is also associated with abnormalities in the lens cytoskeleton and epithelial cell junctions. A total of 55 sequence variants in the gene underlying the X-linked dominant NHS have been identified. Affected men have dense nuclear cataracts and frequently have microcornea, whereas heterozygous women show sutural cataracts with microcornea, craniofacial dysmorphism, nystagmus, strabismus and dental anomalies59 60(table 1).
Studies in various animal models have identified several genes and their pathways linked to lens defects and cataract that are good candidates for examining human patients; for example, iSyTE (https://research.bioinformatics.udel.edu/iSyTE/ppi/) has impacted many cataract gene discoveries both in human and animal models. Recently, Siddam et al61 have shown that deficiency of Celf1 (RNA binding protein) causes cataract in fish and mouse, and serves as a potential candidate to examine in patients with cataract.
Cataract therapeutics and future direction
Understanding cataractogenesis: latency and ageing
The timing of the appearance of cataract depends on whether it is due to a harmful mutation or primarily due to accumulated biomolecular damage. Both can be described as accumulated cataractogenic load, manifesting as cataract either during infancy or at other life stages. Genetic, environmental and stochastic factors all contribute to ageing of the lens and presbyopia at the end of the fourth decade of life is evidence of this. Presbyopia is both a consistent, universal, totally penetrant phenotypic change in the ageing human lens. In line with ageing hypotheses, treatment with antioxidants such as a lipoic acid derivative can reverse presbyopia in mouse models.62 Indeed, Nacety cysteine derivatives are also effective cataract-reversing molecules in other animal models, indicating that cataract, though a histopathological endpoint, is not necessarily irreversible if treatment and timing are optimised.63 64 For each individual, the manifestation of their cataract will depend on their accumulated cataractogenic load due to various genetic and environmental insults. This explains why the age of onset for age-related cataract is so variable and why for congenital cataract the age of onset is more consistent. This can be interpreted as representing a threshold.65
The lens relies on biomolecular longevity66 67 because of the lifelong retention of its components and particularly the proteins. The lens continues to grow throughout life and as such bimolecular integrity is integral to its lifelong functions.68 It is important to recognise that lens metabolism changes as the tissue ages, evidenced by the tissue response to growth factors69 by metabolite accumulation (kynurenine derivatives,70 71 by the gradual reduction in glutathione levels72 and of course the age-dependent increased levels in racemization/isomerisation73 and advanced glycation end-modified lens proteins.74 75 The accumulation of polypeptides as a result of the proteolytic modification of lens proteins can also have beneficial and detrimental effects.76 It is important to appreciate this concept when considering pharmaceutical/small molecule interventions to either arrest or even reverse cataract.
Small molecules
Cataracts are a major burden on public health, and effective therapeutic approaches are yet to be established for preventing or mitigating the process. Lens opacity triggers due to malfunction of alpha-crystallins, when they lose their chaperone properties either due to genetic basis or ageing. Due to their reduced functional capacity, crystallin tends to misfold the protein and aggregate into amyloids.77 Several studies have suggested few compounds that can reverse the light scattering lens aggregates in vitro and in animal studies.
Recently, Makley and colleagues78 identified a small molecule, ‘compound 29’, which binds to several members of the small heat shock protein family including the alpha-crystallins. This compound, an oxysterol, was shown to improve lens transparency in the mouse model and able to restore the protein solubility in the lenses of aged mice in vivo and in human lenses in vitro. These findings suggest an approach to treating cataracts by stabilising alpha-crystallins.
Another group identified variants in the LSS gene in two autosomal-recessive congenital cataract families. The LSS gene encodes lanosterol synthase, a key enzyme in the cholesterol biosynthesis pathway, is highly expressed in the lens and has water-lipid solubility properties. Lanosterol treatment has shown to reverse crystallin aggregation in vitro. It was also suggested to improve lens transparency in rabbits and dogs with naturally occurring cataract. Due to its solubility properties, lanosterol has been suggested as a possible topical eye-drop therapy for cataract.79 80 although recent studies have cast doubt on the efficacy of the identified oxysterols lanosterol and 25-hydroxy cholesterol to treat cataract.81 The chemical chaperone 4-phenylbutyrate was used in mouse lenses to improve lens function in the genetic model of a cataract-causing connexin mutation (Cx50D47A) with limited success.82 The search for small molecule inhibitors has been a consistent research goal over the decades with promising effects obtained with molecules such as pantethine,83rosmarinic acid and polyherbal preparations84 and multifunctional antioxidants.85 Those that have shown the most promise are those that have met the challenge of reducing cataractogenic load and this is therefore an important mechanistic focus for future work.
Cellular therapy
Lin et al have developed a method that enables lens regeneration from endogenous stem cells to treat cataract, where they removed cataract lens in mammals and human infants while preserving lens capsule and lens epithelial cells86 In the case of inherited cataracts, this method deploys further gene editing using CRISPR/Cas9 technology in order to rectify the genetic mutation in the regenerated lens.
However, small molecule therapy, for example using eye drops, seems to remain the most pragmatic solution in developing parts of the world, where immediacy and cost-effectiveness is key. The more invasive and time-consuming stem cell therapy approaches are apt in cases where gene editing is essential to correct the mutant gene, for example, inherited cataracts.
Conclusions
The identification of genetic variants causing congenital cataract has not only improved our understanding of the pathogenesis of infantile cataract, the most frequently treatable cause of blindness in childhood, but also its more common counterpart, adult-onset cataract. For example, LIM2, EPHA2 and TDRD7 that have variants described in both congenital and age-related cataract. It is thus pivotal to identify genetic variations associated with a high risk of developing cataract. This may lead towards new strategies for the prevention of cataract or mitigating the progression of early lens opacity, thus reducing the global huge demand for surgery. There is immense potential in cataract research to elucidate the relationship between neurological and vascular diseases.87
References
Footnotes
Contributors VB: conceived, wrote and provided critical revision of the manuscript. KF, AM, RQ and MM: provided critical revision of the manuscript. MG and VB: produced the figures.
Funding Supported by grants from Rosetree Trust (A2223), the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, Moorfields Eye Hospital Special Trustees, and Moorfields Eye Charity, The Wellcome Trust (099173/Z/12/Z).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.