Article Text

Abstract
Background Ectopia lentis refers to dislocation or subluxation of the crystalline lens. Fibrillin-1, encoded by FBN1, is an important microfibrillar structural component that is specifically required for the suspensory ligament of the lens. FBN1 mutations may cause abnormal structure of microfibrils and has been associated with a broad spectrum of clinical phenotypes. In this study, we characterised a Chinese dominant family with late-onset isolated ectopia lentis caused by a novel missense FBN1 mutation.
Methods Eight family members, including four patients with suspected isolated ectopia lentis, were recruited from Shanghai. Clinical data and family history of the proband and other affected family members were collected. Ophthalmic examination, systemic examination and echocardiography were performed. Whole exome sequencing and Sanger sequencing were used to detect potential pathogenic variants.
Results A novel heterozygous missense mutation c.4031 G>A/p.Gly1344Glu in exon 33 of FBN1 was identified. This mutation was detected in all affected family members and led to specific ocular system phenotypes (ectopia lentis, microspherophakia and secondary glaucoma) with minor skeletal involvement (hallux valgus).
Conclusion The novel c.4031G>A mutation in FBN1 is a likely pathogenic mutation for isolated ectopia lentis. Our study expands the spectrum of FBN1 mutations and contributes to better comprehension of genotype-phenotype correlations of ectopia lentis disease.
- lens and zonules
- genetics
- glaucoma
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Ectopia lentis (EL; Online Mendelian Inheritance in Man (OMIM) #129600) refers to dislocation or subluxation of the crystalline lens resulting from suspensory ligaments dysplasia. It may be present as a part of typical syndromic conditions in many connective tissue disorders, especially Marfan syndrome (MFS; OMIM#154700).1 2 On the other hand, it can also be observed as an independent finding without obvious systemic comorbidities.3 Historically the diagnosis would be isolated ectopia lentis (IEL) for patients who have primary displacement and non-syndromic features.1 However, patients with EL and a mutation in FBN1 may be classified into MFS according to the 2010 Ghent criteria, if their mutation has previously been described with aortic dilation/dissection.4 IEL can be either an autosomal dominant or an autosomal recessive trait. The autosomal dominant form of IEL is most commonly caused by mutations in FBN1. In contrast, mutations in ADAMTSL4 have been consistently associated with the autosomal recessive form.5
FBN1 is located at chromosome 15q21.1 and encodes a 320 kDa cysteine-rich glycoprotein named fibrillin, a major structural component of extracellular matrix (ECM) microfibrils.6 7 Fibrillin is an highly reserved, multidomain structural protein found in all types of connective tissue, which contributes to the elasticity and resiliency of a variety of tissues.8 9 Each fibrillin molecule is composed of 47 epidermal growth factor-like (EGF) domains, among which 43 are calcium binding type (cbEGF), and 7 transforming growth factor-β1 binding protein-like (TB) domains.10 11 FBN1 mutations may lead to abnormal structure of microfibrils and has been associated with a broad spectrum of clinical phenotypes.7 According to Universal Mutation Database (UMD-FBN1), more than 3000 mutations have been identified throughout FBN1, with less than 20 being associated with IEL.1 2 6 12–15
In this study, we characterised a Chinese dominant family in which a novel missense mutation c.4031 G>A/p.Gly1344Glu in FBN1 co-segregates with late-onset IEL and additional manifestations of microspherophakia, secondary glaucoma with minor skeletal involvement.
Materials and methods
Patients information and clinical manifestation
The STREGA (STrengthening the REporting of Genetic Association studies) reporting guidelines was used to improve the reliability of the research.15 The patient visited our hospital due to ectopia lentis and secondary glaucoma. Clinical data and family history of the proband and the remaining affected family members were collected. Ophthalmic examination included visual acuity assessment, intraocular pressure (IOP) measurement, slit-lamp examination (SL-D7, Topcon, Tokyo, Japan), dilated examination of lens and fundus, panoramic ophthalmoscope (Daytona (P200T), Optos, Dunfermline, UK), spectral domain optical coherence tomography (SD-OCT) (TR-KT-2913, Heidelberg Engineering GmbH, Heidelberg, Germany), ultrasound biomicroscopy (UBM) (SW-3200, Suoer, Tianjin, China) and axial length measurement (IOL Master, Carl Zeiss Meditec, Jena, Germany). Systemic examination included measurements of height and skeleton. For each patient and mutation-carrying relative, an echocardiography was performed to detect cardiovascular diseases. After informed consents for diagnostic genetic testing were obtained, peripheral blood samples were collected and genomic DNAs (gDNA) were extracted from available individuals. A total of eight members including four suspected IEL patients (I:2, II:1, II:2, II:3) in a Chinese family from Shanghai were recruited.
Whole exome sequencing
Whole exome sequencing (WES) and data analysis were conducted through BGI Co, Ltd (Shenzhen, China). gDNA was extracted according to the manufacturer’s standard procedure (MagPure Buffy Coat DNA Midi KF Kit). gDNA (Quality Control (QC): gDNA concentration ≥20 ng/ul, total DNA ≥1000 ng) was broken into 100 to 500 bp fragments by BGI’s enzyme kit (Segmentase, BGI). The 280 to 320 bp fragments were then collected by magnetic bead. The collection added ‘A’ base at 3’ overhangs after repairing ending, which made sure the fragments could pare ‘T’ base with special adapter, and built single individual DNA library after ligation-mediated PCR and purification (QC: small fragment concentration ≥40 ng/ul, total gDNA ≥1000 ng). The library was enriched 16 to 24 hour (47℃) by array hybridisation (Roche NimbleGen, Madison, USA), followed by elution and post-capture amplification. The elution concentration of fragments should between 10 ng/ul to 50 ng/ul with fragment size of 300 to 550 bp for QC requirements. The products were then subjected to Agilent 2100 Bioanalyzer and BMG to estimate the magnitude of enrichment. The qualified products were pooled and quantified according different library quantities, then the single strand of library products was prepared for circularisation and made DNA nanoball. Finally, sequencing was performed with 2×100 bp paired end reads (PE 100+100) on MGISEQ-2000 (https://www.mgitech.cn/products/instruments_info/13/). QC standards for bioinformatics included deduplication read depth ≥100X, 20X coverage ≥95%, Q20 ≥90%, Q30 ≥85% with sequencing data volume no less than 15G.
Mutation identification
To detect the potential variants in the family, bioinformatics processing and data analysis were performed after receiving the primary sequencing data. Following previously published filtering criteria, a few unqualified sequences were removed using a BGI in-house programme SOAPnuke to generate ‘clean reads’ for further analysis. The clean reads with a length of 90 bp were then aligned to the human genome reference (hg19) using the BWA (Burrows Wheeler Aligner) Multi-Vision software package. PCR duplicates were removed with Picard tools. After alignment, the output files were used to perform sequencing coverage and depth analysis of the target region, single-nucleotide variants (SNVs) and small inversion and deletion (INDEL) calling. Genome Analysis Toolkit (GATK) software was used to detect SNVs and INDELs, which were filtered and evaluated via multiple databases, including OMIM, NCBI, dbSNP, HapMap, 1000 genomes project data set and in-house database of 100 Chinese Han normal controls.16 17 The minor allele frequency (MAF) of the variants were determined using the gnomAD, ExAC, ESP6500, 1000 Genomes and the in-house sequencing data of 100 Chinese healthy adults. Non-synonymous variants with an MAF of 0.01 or less and consistent with the autosomal dominant inheritance were considered as candidate pathogenic mutations. Potential pathogenic effects of the candidate mutations were evaluated by the MutationTaster, Protein Variation Effect Analyzer (PROVEAN) and Scale-Invariant Feature Transform (SIFT) programme. Pathogenicity of the candidate variant was assessed following guideline issued by American College of Medical Genetics and Genomics (ACMG).18 19 The Human Gene Mutation Database was used to identify mutations reported in previous publications.
Sanger sequencing verification
After targeted sequencing, the FBN1 G1344E mutation and other potential pathogenic variants identified in the proband and the remaining affected family members were verified by Sanger sequencing. A 448 bp fragment was amplified by PCR using the following primers: 5’-AACATACCTGTACAAGTGAAGC-3’ (forward) and 5’-ACATTTGTGCTGAGCCTTTTTC-3’ (reverse). Sequence was compared with the FBN1 reference sequence (NM_000138.4) to identify mutations.
Results
Pedigree and detail clinical manifestations
The pedigree of the family was drawn in figure 1. A novel heterozygous missense mutation (c.4031 G>A/p.Gly1344Glu) in exon 33 of FBN1 was detected following an autosomal-dominant mode. (figure 2) The proband II:1 is a 49-year-old male with normal height (181 cm) and did not fulfil the revised Ghent criteria for Marfan syndrome.4 He had bilateral angle-closure glaucoma and underwent Nd:Yag peripheral iridotomy several years ago. Antiglaucoma medications were used every day. Since the IOP could not be controlled by previous medical management, he came to our hospital for advanced treatment. He had decreased visual acuity (OD: 0.7, OS: 0.2), increased IOP (OD: 18 mm Hg, OS: 49 mm Hg), lenticular dislocation with cataract, microspherophakia, shallow anterior chamber, peripheral iris deformation and myopia (OD: −4.75D, OS: −3.50D) in both eyes. (figure 3) It is noteworthy that he has a shorter axial length (OD: 20.18 mm, OS: 20.49 mm), same as his mutation-carrying mother (I:2), younger sister and brother (II:2, II:3). Supratemporal ciliary body cysts were found in his left eye during pars plana vitrectomy and scleral intraocular lens fixation surgery. A prophylactic laser procedure was performed during the surgery. There are no malformations in his cardiovascular system by ultrasound examination. He has minor skeletal involvement of hallux valgus without affecting the walking function. (figure 4) All affected members in this family manifested similar clinical symptoms, mainly in the ocular system with minor skeletal involvement. (eg, patient I:2, II:2 and II:3, view online supplementary files 1-3) None of them had cardiovascular malformation. In ocular system, all affected members had EL because of suspensory ligaments laxity, microspherophakia, shallow anterior chamber, myopia, increased IOP and decreased axial length. The main skeletal abnormality is hallux valgus, with no arachnodactyly, scoliosis, joint hypermobility or other malformations observed in any of the affected members. At the time this study was performed, III:1 and III:2, two family members in the third generation carrying FBN1 G1344E mutations, have only hallux valgus phenotype with no ocular manifestations. Considering the related disease had a late onset, IEL may appear in their older ages, though further follow-up and observation are necessary to verify the clinical manifestations of this mutation. The detailed clinical information of the family members including the proband was shown in table 1.
Supplemental material
Supplemental material
Supplemental material
Clinical detail of the family members

The pedigree of the family. Square indicates male and circle indicates female. Dark symbol means affected member with ocular and hallux valgus phenotype. Grey symbol means affected member merely with hallux valgus phenotype. I to IV means generations and 1 to 3 means the ID number of each patient. The arrow points to the proband. The genotype information of c.4031 G>A were marked, heterozygous in all affected members and not found in all unaffected members. WT, wild type.

A novel missense mutation (c.4031 G>A/p.Gly1344Glu) in exon 33 of FBN1 was identified. (a) Sanger sequence of unaffected family member. (b) The heterozygous c.4031 G>A mutation of proband.

Ophthalmologic symptoms of the proband (II:1). (a) Slit-lamp examination. The crystalline lentis was dislocation results from suspensory ligaments dysplasia in the left eye. (b) UBM examination shows shallow anterior chamber, peripheral iris deformation, anterior chamber angle crowding and trabecular meshwork blocking of left eye. (c) Panoramic ophthalmoscope and (d) SD-OCT show normal fundus images. SD-OCT,spectral-domain optical coherence tomography; UBM, ultrasound biomicroscopy.

Systemic symptoms of the proband II:1. The patient has minor skeletal involvement of hallux valgus.
Variant of FBN1 identification and Sanger validation
345 candidate variations were remained for further screening after automated filtering. According to semi-automatic mutation screening and interpretation, a total of 12 and 21 autosomal dominant inherited variations were found to be related to patient’s ocular and skeletal system, respectively. Based on the clinical manifestations, FBN1 p.G1344E (c.4031 G>A) mutation was identified as the only candidate pathogenic mutation that related to the patient’s ectopia lentis phenotype. Co-segregation of this variant and the disease phenotype was confirmed within the family members by PCR amplification and Sanger sequencing (heterozygous in all affected members and not found in all unaffected members).
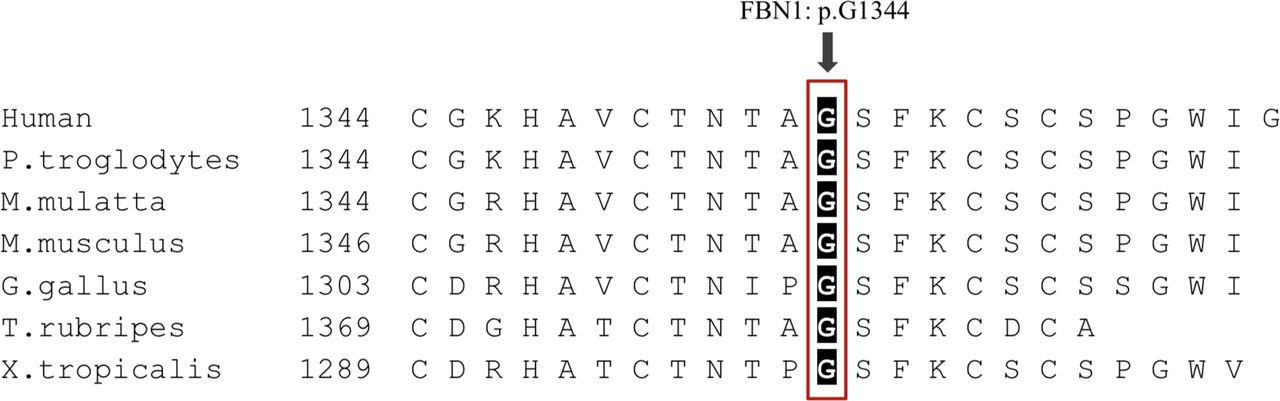
The c.4031 G>A mutation changed codon GGA to GAA in exon 33, substituting an evolutionarily conserved amino acid glycine with glutamic acid in a cb-EGF domain of FBN1 (Phylop score: 5.731; PhastCons score: 1). (figure 5) Based on the ACMG guideline, the p.G1344E variant should be classified as likely pathogenic as it meets with two moderate (PM1 + PM2) and two supporting (PP2 + PP4) criteria. (PM1: located in a mutational hotspot and/or critical and well-established functional domain without benign variation; PM2: absent from controls (or at extremely low frequency if recessive) in gnomAD, ExAC, ESP6500, 1000 Genomes and the in-house sequencing data of 100 Chinese healthy adults; PP2: missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease; PP4: patient’s phenotype or family history is highly specific for a disease with a single genetic aetiology).19 Furthermore, p.G1344E in FBN1 was predicted to be disease-causing by MutationTaster (prob: 0.999; Disease Causing), PROVEAN (score: −7.569; Deleterious) and SIFT (score: 0.999; Probably Damaging), supporting that this mutation was likely pathogenic and associated with the disease phenotype.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Multispecies sequence alignment showing the evolutionarily conserved residues of p.Gly1344 in FBN1.
Discussion
IEL results from instability or rupture of the ciliary zonule, which span the narrow gap between the ciliary epithelium and lens equator.9 Fibrillin microfibrils are specifically required for the suspensory ligament of the lens.12 FBN1 is widely expressed in zonules, ligaments, cartilage, tendon, large arteries and other tissues.20 Fibrillin-1, encoded by FBN1, is an important calcium-binding microfibrillar structural component of ECM that contribute to the integrity and function of all extensible connective tissues.8 12 21 22
FBN1-related disorders include EL, MFS, MASS (mitral valve prolapse, aortic enlargement, skin and skeletal findings) syndrome, autosomal dominant Weill-Marchesani syndrome, acromicric and geleophysic dysplasias.22 Although the phenotypes of these diseases are quite distinct, the major manifestations of these diseases involve ocular, skeletal and cardiovascular system. To date, thousands of mutations have been identified in FBN1, while missense mutations are the most frequent type.23 It has been revealed that cysteine substitution in cbEGF domains and missense mutations in 5’ region have a higher incidence of combined EL.24 25 Since some patients had been reclassified as MFS if the mutation is subsequently described in association with aortic dilation/dissection, less than 20 FBN1 mutations causing IEL have been reported.1 3 6 15 26–34 Two recently IEL reported cases were recommended further aortic disease surveillance to identify or exclude Marfan syndrome because the patients were too young.31 32 Intriguingly, with just a few exceptions, nearly all reported mutations causing IEL were cysteine substitutions that occurred in different exons of FBN1.33 The c.4031G>A/p.Gly1344Glu mutation described in this study is the first non-cysteine-substitution mutation identified in exon 33.
The c.4031G>A mutation mainly caused ocular complications in the affected family members. Considering the related disease had a late onset, although not all mutation-carrying members developed symptoms (III:1, III:2), IEL may appear in their older ages. (Representative case: patient III:2, view online supplementary file 4) It has been reported that the median age of diagnosis of IEL caused by FBN1 mutation was 35 years old.6 In our study, the diagnosis ages of the three affected members were 48 (II:1), 40 (II:2) and 41 (II:3) years old. Our patients apparently had increased risk of developing secondary angle-closure glaucoma, mainly due to crystalline lens subluxation, anterior chamber angle crowding and trabecular meshwork blocking. The shorter axial globe length might advance negative effects on the development of glaucoma.35 It cannot be excluded that some of the EL patients might develop additional features of Marfan syndrome in their adult life. However our patient is older than 20 years old and does not meet the revised Ghent criteria.1 The affected member I:2 is a 72-year-old female without any cardiovascular or other skeletal manifestation. Although she has already lost visual acuity because of glaucoma, it’s reasonable to speculate that mutation in this region was predominantly associated with ocular system phenotypes and less likely to have aortic disease. This specific p.G1344E amino acid substitution might limit the phenotype and severity.
Supplemental material
Based on previous reports and the clinical symptoms of the affected members, we believe that the novel FBN1 mutation identified in this study has a deleterious nature and is the likely cause of IEL for the following reasons: First, heterozygous p.G1344E mutation co-segregated with the EL phenotype in four affected individuals (I:2, II:1, II:2, II:3), and the intrafamilial segregation pattern is consistent with the dominant inheritance. Second, the MAF of p.G1344E was extremely low, absent in the gnomAD, ExAC, ESP6500, 1000 Genomes databases and 100 ethnically-matched normal controls. Third, the p.G1344E mutation changes an evolutionarily conserved amino acid of FBN1 and is unanimously predicted to be disease-causing by computational analysis tools MutationTaster, PROVEAN and SIFT. Fourth, based on the ACMG guideline, the p.G1344E variant should be classified as likely pathogenic as it meets with two moderate (PM1 + PM2) and two supporting (PP2 + PP4) criteria. In summary, this FBN1 mutation is consistent with the previously reported autosomal dominant inheritance of IEL.
There are limitations to our study. Though by WES we identified the p.Gly1344Glu variant in FBN1 as the likely pathogenic causes for the ectopia lentis in the family, our data cannot completely rule out the possibility that a cryptic mutation in tight linkage (in intronic regions of FBN1 not covered by WES, for example) may be present. Within the family, there are two p.G1344E mutation-carrying individuals (III:1 and III:2) with no ocular phenotypes. Considering the onset of the ectopia lentis in this family is rather late and individuals III:1 and III:2 were still relatively young (<30 years), further follow-up and observation are necessary to verify the clinical manifestations of the p.G1344E mutation. Alternatively, however, it is also possible that a second unidentified modifier mutation in certain family members may explain the heterogeneity of the phenotype. Further functional studies are needed to assess the effect of the p.G1344E mutation in the next step.
Conclusion
In conclusion, the novel heterozygous missense mutation c.4031G>A (p.Gly1344Glu) in exon 33 of FBN1 is a likely pathogenic mutation for IEL. Our study expands the spectrum of FBN1 mutations and contributes to better comprehension of genotype-phenotype correlation of IEL disease.
Acknowledgments
The authors would like to thank the patients who participated in this study and thereby made this work possible. The authors would like to thank Ling Li (Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine), Jie Yang (Shanghai Jiao Tong University School of Medicine) and Pan Xiao (Changshu No.2 People’s Hospital) for their assistance in implementing the study protocol.
References
Footnotes
Contributors ZYW and PG takes responsibility for the integrity and the accuracy of the data analysis. ZYW and PG contributed to the study design and collected the samples. YY analysed the data, wrote the first draft of the manuscript and edited the subsequent revisions. YLZ and TTY contributed to the study design and oversaw the implementation of the study protocol. HP reviewed, edited and approved the final manuscript. All authors have read and approved the manuscript.
Funding This project was supported by the National Key R&D Program of China (2018YFC1106100, 2018YFC1106101), the National Natural Science Foundation of China (81770934 & 81870687 & 81702781), Shanghai Municipal Education Commission - Gaofeng Clinical Medicine Grant Support (20181810), Clinical Research Promotion Program of Ninth People's Hospital Affiliated to Medical College of Shanghai Jiao Tong University (JYLJ032), Shanghai Smart Medical Special Research Project (2018ZHYL0221) and the Research Grant of the Shanghai Science and Technology Committee (17DZ2260100).
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval This study complied with the tenets of the Declaration of Helsinki and was approved by the Ethics Committee of the Shanghai Ninth People’s Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. The whole exome sequencing data used and/or analysed during the current study are available from the corresponding author on reasonable request.
Linked Articles
- At a glance