
Abstract
Congenital motor nystagmus (CMN) is characterized by early-onset bilateral ocular oscillations. To identify the disease locus for autosomal dominant CMN in a Chinese family 86001, clinical data, including slit lamp and funduscopic examination and blood samples were collected from family. Genomic DNA was prepared from leukocytes, and a genome-wide linkage scan was performed using 382 polymorphic microsatellite markers and two-point linkage analysis using the logarithm of odds (LOD) score method as implemented in the LINKAGE program package. Maximum two-point scores were calculated using ILINK, and LINKMAP was used for multipoint analysis. All nine affected individuals in the family showed typical phenotypes for CMN. Maximum two-point LOD scores (3.61 at θ=0) were obtained with D1S2619, D1S2877 and D1S2622.The 24.6 cM (28.07 Mb) linked region is flanked by markers D1S218 and D1S2655, placing the disease locus on chromosome 1q25.2–1q32.1. Multipoint analysis confirmed linkage to the region of D1S218 and D1S2655 with Maximum two-point scores of 3.61. The linkage interval overlaps with that of a newly reported CMN locus on 1q31–q32.2 and narrows down the linked region to 5.90 cM (5.92 Mb). This study confirms and refines a novel locus for autosomal dominant CMN to chromosome 1q31.3–q32.1 (5.90 cM) and demonstrates its presence in the Chinese population.
Similar content being viewed by others


Introduction
Nystagmus is rhythmic and repetitive oscillation of the eyes, usually conjugate and horizontal. The prevalence of nystagmus in the general population was estimated to be 1 in 1000 to 1 in 1500.1 Nystagmus is usually associated with other ocular or systemic diseases, such as congenital cataract, albinism, Leber congenital amaurosis, aniridia, cone or cone–rod dystrophy, macular coloboma and optic nerve dysplasia that significantly impair vision early in life.2 However, in rare cases, nystagmus occurs in the absence of other diseases, termed congenital motor nystagmus (CMN) or idiopathic congenital nystagmus,2, 3 in which case the primary defect presumably lies with ocular motor control regions of the brain rather than the normal anatomic ocular structures.
CMN is genetically heterogeneous and can be transmitted as autosomal dominant (NYS2, MIM 164100, 6p12; NYS3, MIM 608345, 7p11; NY4, MIM 193003, 13q31–q33; and a novel CMN locus 1q31–q32.24), autosomal recessive (MIM 257400) or X-linked (NYS1, MIM 31700, Xq26–q27; NYS5, MIM 300589, Xp11.4–11.3; and NYS6, MIM 300814, Xp22.3) trait. To date, at least seven genetic loci for CMN have been suggested2, 3, 4, 5, 6, 7, 8 but only one gene has been identified, FRMD7, for an X-linked form of CMN.9 Therefore, there are probably other genes remaining to be discovered in CMN.
Recently, a novel locus for autosomal dominant CMN has been mapped to 1q31–q32.24 in a Chinese family. In search for new genes responsible for CMN, we recruited one non-consanguineous Chinese family with CMN. In this study, we have mapped autosomal dominant CMN in a Chinese family to chromosome 1q25.2–1q32.1, a 24.6 cM interval flanked by markers D1S218 and D1S2655. This locus confirmed our previous finding of a novel CMN locus at 1q31–q32.2 and further narrowed down the linkage region to1q31.3–q32.1, a 5.90 cM (5.92 Mb) region with 48 genes.
Materials and methods
Family ascertainment
Family 86001 with autosomal dominant CMN was recruited from the Pediatric & Genetics Clinic of the Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China. This study was approved by Institutional Review Board of Zhongshan Ophthalmic Center and National Eye Institute of NIH. Written informed consent conforming to the tenets of the Declaration of Helsinki and following the Guidance of Sample Collection of Human Genetic Diseases (863-Plan) by the Ministry of Public Health of China was obtained from the participating individuals or their guardians before the study. Diagnostic criteria for CMN are the same as we described in our previous study.4 Genomic DNA was prepared from leukocytes of peripheral venous blood.
Genotype analysis and linkage analysis
Genotyping for all participating family members was performed using 5′ fluorescently labeled microsatellite markers. A genome-wide linkage scan was performed with 382 highly polymorphic fluorescent markers from the ABI PRISM Linkage Mapping Set MD-10 (Applied Biosystems, Foster City, CA, USA). Multiplex PCR was carried out in a GeneAmp PCR System 9700 thermocycler (Applied Biosystems). Briefly, each reaction was carried out in a 5 μl mixture containing 20 ng genomic DNA. Initial denaturation was carried out for 5 min at 95 °C, followed by 10 cycles of 15 s at 95 °C, 15 s at 55 °C, and 30 s at 72 °C, and then 20 cycles of 15 s at 89 °C, 15 s at 55 °C, and 30 s at 72 °C. The final extension was performed for 10 min at 72 °C. PCR products for a panel subset of markers from each subject were pooled and mixed with HD-400 size standards (Applied Biosystems). Alleles were separated using an ABI 3130 DNA analyzer (Applied Biosystems) and then assigned using GeneMapper (version 4.0; Applied Biosystems). Two-point linkage analyses were performed using the MLINK from the LINKAGE program package.10, 11 Maximum logarithm of odds (LOD) scores were calculated using ILINK, and LINKMAP was used for multipoint analysis. Autosomal dominant congenital idiopathic nystagmus (CIN) was analyzed as a fully penetrant trait with an affected allele frequency of 0.0001. Marker allele frequencies were estimated from 50 control chromosomes. The marker order and distances between the markers were obtained from the Généthon database. For fine mapping around the candidate linkage region, additional markers were selected according to the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/unists/) map. An M13-tailed primer PCR method was used to genotype the additional markers.12 Haplotypes were generated using the Cyrillic 2.1 program (Cyrillic Software, Wallingford, Oxfordshire, UK) and confirmed by inspection. The criteria for establishing linkage have been described previously.13
Candidate gene screening
A candidate gene known to cause ocular disease in the linked region, CRB1, was analyzed by Sanger Sequencing. PCR amplifications were carried out in 10 μl reactions containing 40 ng genomic DNA. Touchdown PCR amplification consisted of a denaturizing step at 96 °C for 5 min, followed by decreasing the annealing temperature from an initial 64 °C in a stepwise fashion by 0.5 °C every second cycle for 15 cycles, then an annealing temperature of 57 °C for 20 cycles and finally a prolonged elongation step at 72 °C for 10 min. Sequencing results were assembled using an ABI PRISM 3130 automated sequencer (Applied Biosystems) and analyzed with Seqman software (DNAStar Lasergene 8, Madison, WI, USA) and Mutation Surveyor (SoftGenetics, State College, PA, USA). A missense mutation is considered to be damaging as judged by the position specific independent counts algorithm of Polyphen214 and SIFT score (which distinguishes tolerated variants from those that are not tolerated) equal to or <0.05, as well as being absent from 192 ethnically matched control chromosomes. Splicing changes were predicted using Automated Splice Site Analyses (https://splice.uwo.ca/).
Results
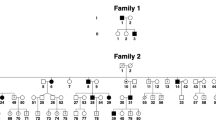
Family 86001 was from the Guangdong province, China. Fifteen individuals, including nine affected and six unaffected participated in this study. In the family, four males and five females are affected with male-to-male transmission, suggesting an autosomal dominant pattern of inheritance (Figure 1). All patients had nystagmus since infancy. Clinical examination of all patients demonstrated horizontal pendular nystagmus, clear corneas, normal irises without pigmentary anomalies, transparent lenses and normal fundi and foveal reflexes. All patients showed normal color vision in recognizing color photos of Ishihara plates. None of the patients had photophobia or night blindness. No systemic abnormalities were observed in the patients.

Pedigree and haplotype construction of family 86001. The haplotypes are composed of nine adjacent microsatellite markers from chromosome 1q25.2–1q32.1. Long black bars indicate the chromosome region with disease allele that is shared by patients, while the other bars represent normal alleles.
Linkage to three known loci for autosomal dominant CMN, including NYS2 (MIM 164100, 6p12),5 NYS3 (MIM 608345, 7p11) and NYS4 (MIM 193003,13q)7 were initially excluded by genotyping closely flanking markers and subsequent two-point linkage analysis. Then a genome-wide linkage scan was performed for the family, yielding LOD scores greater than 1.0 for markers D1S238, D1S413 and D4S406 at θ=0. Of these markers, D4S406 had closely flanking markers D4S1572 and D4S402, yielding LOD scores of minus infinity (the distances with D4S406 is 9.5 and 7.7 cM, respectively) and haplotype analysis excluding true linkage to the region. In contrast, D1S238 and D1S413 are adjacent markers in the MD-10 mapping set, yielding LOD scores of 3.57 and 2.11 at θ=0, respectively.
Fine mapping with closely spaced markers was carried out in order to refine the linked region around D1S238 and D1S413 further. Maximum LOD scores of 3.61 were obtained for both markers D1S2619, D1S2877 and D1S2622 at θ=0 (Table 1). Haplotype analysis further supports the results of the linkage analysis (Figure 1) recombination between D1S218 and D1S2619 in unaffected individual 16 sets indicates the proximal boundary of the linked region, while recombination between D1S2622 and D1S2655 in affected individual 13 sets indicates the telomeric boundary. Eight of 9 markers between D1S218 and D1S2655 gave positive LOD scores greater than 2.0 at θ=0 and all patients shared a haplotype comprising these 9 markers. These results strongly suggest that the disease gene is located in a 24.6 cM region (28.06 Mb) between markers D1S218 and D1S2655 on chromosome 1q25.2–1q32.1. Multipoint linkage analysis carried out with overlapping sets of three markers from the Genethon database confirmed linkage to this 24.6 cM (28.07 Mb) region located between markers D1S218 and D1S2655 with maximum LOD score of 3.61 (Figure 2).

Multipoint linkage analysis in family 86001. The genetic distances (in cM) are taken from the Genethon database chromosome 1 map. A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
In this study, autosomal dominant CMN in a Chinese family was mapped to a 24.6 cM region (28.06 Mb) between markers D1S218 and D1S2655 on chromosome 1q25.2–1q32.1. This region overlaps that of a recently identified locus for autosomal dominant CMN in chromosome 1q31.3–q32.2 between D1S2816 and D1S2692, a 12.1 cM (11.39 Mb) region.4 The overlapping linked intervals shared between the current study and that of Xiao et al.4 resides between D1S2816 and D1S2655, a 5.90 cM (5.92 Mb) region on 1q31.3–q32.1 (Figure 3). Although both Chinese families are of Han nationality and are from Guangdong province, they are unrelated and live in different areas separated by a distance of about 450 km.

Diagram of the chromosome 1 showing the critical region for autosomal dominant CMN. a and b denotes linked regions in this study and Xiao (2011), respectively. The linkage interval shared by the two families is between D1S2816 and D1S2655, a 5.90 cM (5.92 Mb) region on 1q31.3–q32.1.
There are 48 genes situated in the common interval between D1S2816 and D1S2655. CRB1 was selected for analysis based on nystagmus that is usually associated with Leber congenital amaurosis. We sequenced all coding exons but did not identify any potential disease causing mutations in this gene. Neither did exome sequencing in the family described by Xiao et al.,4 mapped to this region, detect causative mutations. Further analysis of the candidate genes in the shared linkage interval may lead to identification of the gene responsible for CMN. Identification of additional loci and refinement of known loci will provide important value toward the understanding the genetic basis of CMN, while eventual elucidation of the molecular mechanisms of CMN will provide insight into the nature of ocular motor control.
References
Self, J. & Lotery, A. A review of the molecular genetics of congenital Idiopathic Nystagmus (CIN). Ophthalmic. Genet. 28, 187–191 (2007).
Cabot, A., Rozet, J. M., Gerber, S., Perrault, I., Ducroq, D., Smahi, A. et al. A gene for X-linked idiopathic congenital nystagmus (NYS1) maps to chromosome Xp11.4-p11.3. Am. J. Hum. Genet. 64, 1141–1146 (1999).
Kerrison, J. B., Vagefi, M. R., Barmada, M. M. & Maumenee, I. H. Congenital motor nystagmus linked to Xq26-q27. Am. J. Hum. Genet. 64, 600–607 (1999).
Xiao, X., Li, S., Guo, X. & Zhang, Q. A novel locus for autosomal dominant congenital motor nystagmus mapped to 1q31-q32.2 between D1S2816 and D1S2692. Hum. Genet. 131, 697–702 (2011).
Kerrison, J. B., Arnould, V. J., Barmada, M. M., Koenekoop, R. K., Schmeckpeper, B. J. & Maumenee, I. H. A gene for autosomal dominant congenital nystagmus localizes to 6p12. Genomics 33, 523–526 (1996).
Klein, C., Vieregge, P., Heide, W., Kemper, B., Hagedorn-Greiwe, M., Hagenah, J. et al. Exclusion of chromosome regions 6p12 and 15q11, but not chromosome region 7p11, in a German family with autosomal dominant congenital nystagmus. Genomics 54, 176–177 (1998).
Ragge, N. K., Hartley, C., Dearlove, A. M., Walker, J., Russell-Eggitt, I. & Harris, C. M. Familial vestibulocerebellar disorder maps to chromosome 13q31-q33: a new nystagmus locus. J. Med. Genet. 40, 37–41 (2003).
Liu, J. Y., Ren, X., Yang, X., Guo, T., Yao, Q., Li, L. et al. Identification of a novel GPR143 mutation in a large Chinese family with congenital nystagmus as the most prominent and consistent manifestation. J. Hum. Genet. 52, 565–570 (2007).
Tarpey, P., Thomas, S., Sarvananthan, N., Mallya, U., Lisgo, S., Talbot, C. J. et al. Mutations in FRMD7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat. Genet. 38, 1242–1244 (2006).
Lathrop, G. M. & Lalouel, J. M. Easy calculations of lod scores and genetic risks on small computers. Am. J. Hum. Genet. 36, 460–465 (1984).
Schaffer, A. A., Gupta, S. K., Shriram, K. & Cottingham, R. W. Avoiding recomputation in genetic linkage analysis. Hum. Hered. 44, 225–237 (1994).
Barkley, N. A., Dean, R. E., Pittman, R. N., Wang, M. L., Holbrook, C. C. & Pederson, G. A. Genetic diversity of cultivated and wild-type peanuts evaluated with M13-tailed SSR markers and sequencing. Genet. Res. 89, 93–106 (2007).
Lander, E. & Kruglyak, L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat. Genet. 11, 241–247 (1995).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Acknowledgements
We thank all patients and controls for their participation. This study was supported by the National Natural Science Foundation of China (30800615), the National Science Fund for Distinguished Young Scholars (30725044) and the Fundamental Research Funds of State Key Laboratory of Ophthalmology.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Li, L., Xiao, X., Yi, C. et al. Confirmation and refinement of an autosomal dominant congenital motor nystagmus locus in chromosome 1q31.3–q32.1. J Hum Genet 57, 756–759 (2012). https://doi.org/10.1038/jhg.2012.103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2012.103
