
ABSTRACT
A tumor vasculature is highly unstable and immature, characterized by a high proliferation rate of endothelial cells, hyper-permeability, and chaotic blood flow. The dysfunctional vasculature gives rise to continual plasma leakage and hypoxia in the tumor, resulting in constant on-sets of inflammation and angiogenesis. Tumors are thus likened to wounds that will not heal. The lack of functional mural cells, including pericytes and vascular smooth muscle cells, in tumor vascular structure contributes significantly to the abnormality of tumor vessels. Angiopoietin-1 (Ang1) is a physiological angiogenesis promoter during embryonic development. The function of Ang1 is essential to endothelial cell survival, vascular branching, and pericyte recruitment. However, an increasing amount of experimental data suggest that Ang1-stimulated association of mural cells with endothelial cells lead to stabilization of newly formed blood vessels. This in turn may limit the otherwise continuous angiogenesis in the tumor, and consequently give rise to inhibition of tumor growth. We discuss the enigmatic role of Ang1 in tumor angiogenesis in this review.
Similar content being viewed by others

INTRODUCTION
Normal blood vessels are composed of two distinct cell types: endothelial cells, the most well studied component of blood vessels with regard to cancer angiogenesis, and mural cells. The mature quiescent vasculature of most organs is characterized by extensive coverage by mural cells; in capillaries and smaller vessels, the mural cell component is comprised of pericytes, whereas in larger vessels this role is fulfilled by smooth muscle cells. This investiture of the endothelial tubule by mural cells is thought to play a major role in maintenance of the quiescent state. For new blood vessel formation to occur, the mural cell coating of the preexisting vessel must first be dissociated, followed by matrix degradation and extravascular fibrin deposition, and freeing of endothelial cells to respond to angiogenic signals with proliferation, migration and tubule formation (reviewed in1). Remodeling occurs to prune vessels to fit the needs of the tissue. This is then followed by a maturation phase characterized by investiture of the endothelial tubule with mural cells leading to quiescence of both cell types, with subsequent basement membrane reconstitution, establishment of cell-cell junctional complexes, and stabilization of the vessel. Coordinated regulation of pro- and anti-angiogenic factors is necessary for each stage to ensure the development of a normal, functional vessel.
It is well established that tumors must acquire the ability to stimulate capillary formation to progress from a small localized growth with a limited oxygen and nutrient supply to a well-vascularized enlarged tumor2. The primary driving force for tumor angiogenesis is the combination of the demand for oxygen and nutrients by the growing cancer cells, and the physical limit of the distances for small molecules to diffuse across the stroma between a nearby capillary and the cell making the demand, a physical limit of about 100 mm3. Folkman and colleagues suggested that once the radius of the tumor reaches this limit, hypoxic conditions occur in the center of the cell mass. An `angiogenic switch' then takes place in favor of new blood vessel growth4.
Several different mechanisms have been proposed to lead to vascularization of tumors. Folkman and colleagues suggested that the tumor induces capillary sprouts from surrounding vasculature by altering the local balance of angiogenic promoters and inhibitors2, in a process known as angiogenesis. Alternatively, Yancopolous and colleagues proposed that cancer cells initially encroach upon existing microvessels (co-opting), this being followed by destabilization and regression of the vessels in the center of the tumor mass, and initiation of new capillary growth at the periphery of the tumor5. Moreover, Dvorak and colleagues suggested that a tumor prior to its own expansion would prepare a microvascular network by stimulating angiogenesis in its immediate surrounds, then utilize these vessels in the expansion phase6. A considerable collection of growth factors and cytokines are shown to take part in the modulation of tumor angiogenesis. However, among the most important components of the potential to initiate angiogenesis in a tumor are plasma leakage and hypoxia.
Abnormality of tumor blood vessels
Due to the aberrant expression of angiogenic factors, tumor vessels develop very abnormally, giving rise to a highly dysfunctional vasculature7. Tumor blood vessels are dilated, with uneven diameters and excessive branching and shunts. The tortuous blood flow is inadequate and leads to hypoxia and acidic regions. The vessel walls are also abnormal, characterized by the presence of a large number of endothelial fenestrae and trans-cellular openings, widened intercellular junctions, and a discontinuous basement membrane. Finally, tumor blood vessels are characterized by decreased mural cell investiture8, 9, 10; ultrastructural studies demonstrate that even when mural cells are present, they exhibit abnormal association with the underlying endothelial tubule11, 12. Consequent to this lack of maturation, tumor vessels are highly permeable with significant plasma extravasation13, 14.
As a result of this permeability, Dvorak likened tumors to wounds that do not heal 15. In a wound, plasma leakage is the result of tissue injury, whereas tumor cells secrete vascular permeability factor (VPF), perhaps better now known as Vascular Endothelial Growth Factor (VEGF), which renders vessels hyperpermeable. During wound healing, platelets facilitate generation of the provisional matrix and initiate the wound healing process; platelets are also important in the later stage of wound healing because they produce platelet-derived growth factor (PDGF), a potent mitogen and chemoattractant for precursors of smooth muscle cells and pericytes 16. In contrast, platelets have not been found outside of blood vessels of solid tumors. Moreover, fibrin and fibronectin appear only transiently in wounds that heal normally, being replaced by type I and III collagen in which the density of blood vessels diminishes17. Fibrin and fibronectin persist in tumor stroma, however, probably due to constitutive tumor production of VPF/VEGF, which results in protracted vessel leakage and continuing clotting of extravasated fibrinogen and fibronectin; in wounds, by contrast, vascular permeability is repaired within a few days after injury. Tumors are thus likened to an unending series of wounds that continually initiate healing and angiogenesis but are unable to heal completely.
Angiopoietins and Tie2
Many growth factors are proposed to play a role in both physiological and pathological angiogenesis. One family of vascular regulatory molecules which has been the subject of intense investigation in both physiological and pathological blood vessel generation are the angiopoietins. The Angiopoietin family of growth factors is comprised of four family members that bind to the Tie2 tyrosine kinase receptor with different outcomes. Angiopoietin-1 (Ang1), the main ligand for Tie218, 19, and -420 are agonistic ligands, whereas Angiopoietin-2 (Ang2) and -3 can serve as antagonistic ligands20, 21. Although Ang1 does not stimulate proliferation of endothelial cells 18, in vitro Ang1 can induce endothelial migration22, tubule formation23 and sprouting24, 25, and survival from a variety of apoptotic insults26, 27, 28, 29, suggesting that Ang1 can be a potent pro-angiogenic factor. Transgenic null mutation of the Ang1 gene confirms an angiogenic role for Ang1, as Ang1 null embryos are unable to form a complex vascular network and exhibit decreased vessel support by mural cells19. These results gave the first indication that Ang1 may play a role in recruitment of mural cells to support the primitive endothelial tubule and enhance vessel maturation. Transgenic Ang1 overexpression or systemic adenoviral delivery resulted in increased vascular branching30, 31, 32. Since Ang1 is not a mitogen, the increased vascular branching may arise from reinforcement of VEGF-induced angiogenesis. Indeed, Ang1 has been shown to synergise with VEGF to enhance angiogenesis in the rat aorta model33 and increase vessel density in the corneal implant assay34 and several other in vivo assays35, 36, 37, 38. While overexpression of VEGF alone gives rise to increased vascular branching, the vessels induced by VEGF are leaky32. By contrast, the vessels induced in the presence of Ang1 are not leaky and resist leakage induced by inflammatory agents32, 39, 40. Part of the means by which this resistance to leakiness occurs may be attributed to markedly enhanced pericyte coverage of the nascent vessels.
The role of Ang2 in blood vessel regulation is quite complex. Transgenic overexpression of Ang2 leads to a phenotype essentially the same as that seen in the Ang1 knockout, suggesting that Ang2 serves as an antagonist for Ang121. This prediction has held true in vitro, as Ang2 can prevent Ang1-stimulated effects on endothelial cells including phosphorylation of Tie221 and migration22. Interestingly, however, it has been shown that Ang2 can activate ectopically-expressed Tie2 on fibroblasts22 and can activate endothelial Tie2 at high concentrations41 or when cells are plated on fibrin42 or collagen matrix43. Indeed, the Ang2 knockout demonstrates that while Ang2 is dispensable for embryonic vascular development, Ang2 is required for both the vascular regression and sprouting events involved in postnatal ocular angiogenesis44.
An intimate relationship between angiopoietins and VEGF in angiogenesis was predicted by analysis of Ang1, Ang2, and VEGF expression in cyclical rat ovary angiogenesis. In this study, Ang2 and VEGF mRNA were co-expressed at the front of invading sprouts during active angiogenesis, whereas Ang2 is upregulated and VEGF is downregulated during vessel regression21. While Ang1 mRNA expression is relatively stable throughout the process, the Ang2 to Ang1 ratio is drastically elevated during corpus luteum vessel regression compared to angiogenesis during corpus luteum formation45. These studies lead to the current dogma that Ang1 and VEGF promote angiogenesis and vessel maturation, whereas Ang2 serves to antagonize the mural cell contact induced by Ang1; in the presence of VEGF, angiogenesis ensues while in the absence, vessels regress21. This notion is further supported by analysis of angiogenesis in the pupillary membrane. Ang2 induces proliferation and migration of endothelial cells and stimulates sprouting of new blood vessels when VEGF is present, whereas it promotes endothelial cell death and vessel regression when the activity of endogenous VEGF is inhibited46.
Null mutation of the gene for the Tie2 angiopoietin rereceptor gave rise to a phenotype similar to both Ang1 null and Ang2 transgenic mice47, 48. Since Tie2 is thought to be largely specific to endothelial cells, it has been suggested that Ang1 activates the Tie2 receptor on endothelial cells, resulting in a yet uncharacterized paracrine loop between EC and SMC. As transgenic knockout approaches targeting PDGF-B/bR give rise to vessels that similarly lack sufficient mural cell investiture16, 49, PDGF has been suggested as a candidate for such a paracrine loop but this has yet to be documented experimentally. We and others have recently reported that mesenchymal mural cell precursor cells, smooth muscle cells, and pericytes express Tie250, 51, 52, 53, and that Tie2 levels can be further upregulated on smooth muscle cells53 and pericytes52 by VEGF. Further, Ang1 can induce migration of mural cell precursors51 and VEGF-preconditioned smooth muscle cells53. These data suggest that part of the mechanism of Ang1-induced vessel maturation may be direct stimulatory action on mural cells.
Angiopoietin expression in tumors
Given the importance of angiopoietins in vascular development, it was of interest to determine what role these factors may play in tumor angiogenesis. In general, high levels of Ang2 by tumor or vascular tissues have been documented in a wide variety of highly vascularized tumors such as malignant glioblastoma54, 55, 56, non-small cell lung cancer57, 58, hepatocellular carcinoma 59, 60, 61, 62, gastric carcinoma63, Kaposi's sarcoma and angiosarcoma64, neuroblastoma65 and thyroid tumor66 (see Tab 1). Further, Ang2 expression has been correlated with poor prognosis in NSCLC 67, HCC62, gastric63 and breast68 cancers. In addition, HT29 colon cancer, hepatocellular carcinoma, and MKN-7 gastric cancer cells engineered to overexpress Ang2 demonstrated augmented tumor growth and vessel count compared to vector controls59, 63, 69. In many cases, VEGF overexpression is observed as well, suggesting that destabilization by Ang2 permits VEGF-induced angiogenesis to proceed (see Tab 1). Indeed, VEGF has been reported to upregulate endothelial Ang2 in vitro70 and in vivo71, although in a C6 brain tumor model, Ang2 expression precedes the appearance of VEGF during the initiation of tumor angiogenesis72, 73. Increased expression of Ang2 is thought to play an integral role in both the proposed mechanism of vessel co-option5, 74 as well as sprouting angiogenesis73. Interestingly, Lewis Lung carcinoma or TA3 mammary carcinoma cells transfected with Ang2 result in decreased tumorigenesis, which the authors attributed to an imbalance with VEGF expression that allowed for vessel regression75. It should be noted that Ang2-overexpressing xenograft vessels lacked coverage by mural cells54,63, 71, 75, and an inverse correlation between Ang2 upregulation and pericyte coverage has been observed in human gliomas54, 74, 76. Finally, in a chemically induced skin carcinogenesis model, Ang2 is not expressed in normal skin, but is upregulated at an early stage during papillomagenesis77. Not surprisingly, Ang2 expression has been reported to be upregulated by hypoxia in microvascular endothelial70, 78 and glioma cells in vitro74 and in capillary endothelium in vivo78, 79. Taken together, these results suggest that Ang2 plays an important role in the initiation of tumor angiogenesis, presumably by its ability to antagonize Ang1-induced, mural cell-mediated vessel stabilization.
By contrast, the role of Ang1 in tumor angiogenesis is less clear. Overexpression of Ang1 has been documented in malignant glioblastoma54, 55, neuroblastoma65, non-small cell lung cancer57, and variably in other tumors as well (see Table 1). In addition, in a Hela xenograft model, Ang1 antisense RNA lead to decreased tumor growth and angiogenesis80 and overexpression of Ang1 promoted HeLa tumor angiogenesis81, suggesting that Ang1 may stimulate angiogenesis in that model. Increasing evidence, however, is suggesting a lack of a role for Ang1 in several cancers in a clinical setting. Recently, we reported that breast cancer epithelial cells do not express Ang182; others have confirmed decreased Ang1 expression in breast tumors compared to normal breast tissue83. Similarly, immunohistochemistry studies demonstrated that Ang1 is expressed in normal colonic epithelium, whereas colon tumors lack appreciable Ang1 staining84. Further, in a mouse skin carcinogenesis model, it was shown that Ang1 expression was completely abolished in papillomas compared to normal skin, and that Ang1 was downregulated in mutant ras-bearing keratinocyte cell lines85. In addition, hypoxia, a principal driver of tumor angiogenesis, has been shown to downregulate Ang1 production by glioblastoma cell lines55 and fibroblasts86. These studies suggest a selective loss of Ang1 expression during the progression toward malignancy.
Inhibition of tumor growth b Ang1 overexpression
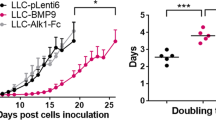
In sharp contrast to the findings from the transgenic studies showing that Ang1 is a promoter of vasculogenesis and angiogenesis during embryonic development, studies using xenograft models demonstrate that ectopic expression of Ang1 in breast82 and colon69 cancer cells results in decreased tumor proliferation and angiogenesis. In addition, Ang1 inhibits colon cancer peritoneal87 and hepatic metastases88. A similar tumor inhibitory role for Ang1 has been observed for squamous cell carcinoma (SCC), as stable overex-pression of Ang1 in A431 xenograft model showed inhibition of tumor growth77, and the K14-HPV16/K14-Ang1 double transgenic developed fewer pre-malignant and tumorigenic lesions than K14-HPV16 parentals 89. These data suggest that, despite its important stimulatory role in embyronic blood vessel formation, Ang1 may exert an inhibitory role for tumor angiogenesis.
What is the mechanism by which the angiogenic factor Ang1 can paradoxically inhibit tumor angiogenesis? Analysis of blood vessels from the Ang1-inhibited tumor models above suggest that the answer may lie in the ability of Ang1 to recruit mural cells to stabilize the blood vessel. Tumor vessels in the Ang1-transfected breast50, colon69, hepatic colon tumor88, and squamous cell77 xenografts mentioned above all demonstrate significantly increased association of pericytes with vessels, suggesting that enforced maturation of blood vessels may functionally inhibit tumor angiogenesis. First of all, the ability of Ang1 to inhibit vascular permeability is thought to be due partly to the enhancement of cell-cell junctions40, as well stabilization of blood vessels by the promotion of mural cell recruitment19,34, 35, 38; it is interesting to note that the absence of pericytes leads to defects in endothelial junction formation16. In addition, peripheral blood vessels in the Ang1-transfected MCF7 tumors were not dilated, in contrast to those in the vector control counterparts50. This Ang1-mediated decline in vessel permeability may decrease the plasma extravasation that creates the permissive, or even stimulatory, environment for further angiogenesis.
Secondly, the presence of mural cells is postulated to be inhibitory for endothelial angiogenic responses. Indeed, several studies90, 91 have demonstrated that actively proliferating endothelium lacks coverage by mural cells, and ultrastructural analysis of breast tumors showed that vessels in areas of low vascular density had greater pericyte coverage than areas of high vascular density92. Further, the loss of pericytes observed in PDGF-B/bR knockouts is concomitant with endothelial hyperplasia16. In addition, mural cell-endothelial cell interactions are reduced following stimulation of angiogenesis93, 94, and the arrival of pericytes coincides with the cessation of vessel growth during wound healing95, suggesting that contact with pericytes leads to quiescence of endothelial cells. In vitro studies by culturing endothelial cells with smooth muscle cells or pericytes using a variety of models corroborate the decreased growth of endothelial cells under these conditions in a fashion that requires cell-cell contact96, 97. Mural cells have been further implicated in the prevention endothelial cell migration98 and sprouting99, and activation of endothelial MT1-MMP100.
Numerous studies have indicated the important role of VEGF in both physiological and pathological angio-genesis. The intimate interaction of VEGF with the angiopoietins leads to the ability to tightly control the angiogenic process. It is thought that the antagonism of Ang1-induced vascular stability by Ang2 leads to dissociation of the mural cell coating to initiate angiogenesis; the ratio of Ang1 to Ang2 is likely to be a critical determinant in this process. In the presence of VEGF, endothelial cells can proliferate, migrate, and form tubules, and in some cases this may act in synergy with Ang1 that is present32, 36, 37. It is tantalizing to suggest that the expression of Tie2 by mural cells and their precursors, and the ability of VEGF to upregulate Tie2 on these cells, renders them responsive to Ang1-mediated migration, leading to investiture of the endothelial tubule with mural cells. This stabilization makes endothelial cells unresponsive to further angiogenic cues and thereby terminates angiogenesis. Ang1-mediated stabilization of tumor blood vessels may therefore be desirable therapeutically to inhibit new vessel formation and thereby arrest tumor growth.
CONCLUSIONS
Researche on anti-angiogenesis as an anti-cancer approach has for some time focused on endothelial cells. It is not until recently that the role of mural cells has drawn more and more attention from cancer researchers, despite the fact that mural cells are an important component of the vascular wall, and their functions have been studied extensively in the cardiovascular field. It is plausible that the anti-angiogenesis effect of vascular stabilization in tumors may lead to new approaches to the development of cancer therapies.
References
Carmeliet P . Mechanisms of angiogenesis and arteriogenesis. Nat Med 2000; 6(4):389–95.
Folkman J . Tumor angiogenesis: a possible control point in tumor growth. Ann Intern Med 1975; 82(1):96–100.
Grote J, Susskind RVaupel P . Oxygen diffusion constants D and K of tumor tissue (DS-carcinosarcoma) and their temperature dependence. Adv Exp Med Biol 1977; 94(3):61–5.
Hanahan DFolkman J . Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996; 86(3):353–64.
Holash J, Maisonpierre PC, Compton D, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999; 284(5422):1994–8.
Brown LF, Guidi AJ, Schnitt SJ, et al. Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin Cancer Res 1999; 5(5):1041–56.
Carmeliet PJain RK . Angiogenesis in cancer and other diseases. Nature 2000; 407(6801):249–57.
Benjamin LE, Golijanin D, Itin A, Pode DKeshet E . Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest 1999; 103(2):159–65.
Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KHAugustin HG . Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res 2000; 60(5):1388–93.
Gee MS, Procopio WN, Makonnen S, Feldman MD, Yeilding NMLee WM . Tumor vessel development and maturation impose limits on the effectiveness of anti-vascular therapy. Am J Pathol 2003; 162(1):183–93.
Abramsson A, Berlin O, Papayan H, Paulin D, Shani MBetsholtz C . Analysis of mural cell recruitment to tumor vessels. Circulation 2002; 105(1):112–7.
Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RKMcDonald DM . Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol 2002; 160(3):985–1000.
Dvorak HF, Nagy JA, Dvorak JT, Dvorak AM . Identification and characterization of the blood vessels of solid tumors that are leaky to circulating macromolecules. Am J Pathol 1988; 133(1):95–109.
Yuan F, Leunig M, Berk DAJain RK . Microvascular permeability of albumin, vascular surface area, and vascular volume measured in human adenocarcinoma LS174T using dorsal chamber in SCID mice. Microvasc Res 1993; 45(3):269–89.
Dvorak HF . Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986; 315(26):1650–9.
Hellstrom M, Gerhardt H, Kalen M, et al. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol 2001; 153(3):543–53.
Tonnesen MG, Feng XClark RA . Angiogenesis in wound healing. J Investig Dermatol Symp Proc 2000; 5(1):40–6.
Davis S, Aldrich TH, Jones PF, et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 1996; 87(7):1161–9.
Suri C, Jones PF, Patan S, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996; 87(7):1171–80.
Valenzuela DM, Griffiths JA, Rojas J, et al. Angiopoietins 3 and 4: diverging gene counterparts in mice and humans. Proc Natl Acad Sci USA 1999; 96(5):1904–9.
Maisonpierre PC, Suri C, Jones PF, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997; 277(5322):55–60.
Witzenbichler B, Maisonpierre PC, Jones P, Yancopoulos GDIsner JM . Chemotactic properties of angiopoietin-1 and -2, ligands for the endothelial-specific receptor tyrosine kinase Tie2. J Biol Chem 1998; 273(29):18514–21.
Hayes AJ, Huang WQ, Mallah J, Yang D, Lippman MELi LY . Angiopoietin-1 and its receptor tie-2 participate in the regulation of capillary-like tubule formation and survival of endothelial cells. Microvasc Res 1999; 58(3):224–37.
Koblizek TI, Weiss C, Yancopoulos GD, Deutsch URisau W . Angiopoietin-1 induces sprouting angiogenesis in vitro. Curr Biol 1998; 8(9):529–32.
KIM I, Kim HG, Moon S-O, et al. Angiopoietin-1 induces endothelial cell sprouting through the activation of focal adhesion kinase and plasmin secretion. Circ Res 2000; 86(9):952–59.
Kwak HJ, So JN, Lee SJ, Kim IKoh GY . Angiopoietin-1 is an apoptosis survival factor for endothelial cells. FEBS Lett 1999; 448(2-3):249–53.
Papapetropoulos A, Garcia-Cardena G, Dengler TJ, Maisonpierre PC, Yancopoulos GDSessa WC . Direct actions of angiopoietin-1 on human endothelium: evidence for network stabilization, cell survival, and interaction with other angiogenic growth factors. Lab Invest 1999; 79(2):213–23.
Kim I, Kim HG, So JN, Kim JH, Kwak HJKoh GY . Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-Kinase/Akt signal transduction pathway. Circ Res 2000; 86(1):24–9.
Kwak HJ, Lee SJ, Lee YH, et al. Angiopoietin-1 inhibits irradiation- and mannitol-induced apoptosis in endothelial cells. Circulation 2000; 101(19):2317–24.
Shyu KG, Manor O, Magner M, Yancopoulos GDIsner JM . Direct intramuscular injection of plasmid DNA encoding angiopoietin-1 but not angiopoietin-2 augments revascu- larization in the rabbit ischemic hindlimb. Circulation 1998; 98(19):2081–7.
Suri C, McClain J, Thurston G, et al. Increased vascularization in mice overexpressing angiopoietin-1. Science 1998; 282(5388):468–71.
Thurston G, Suri C, Smith K, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 1999; 286(5499):2511–4.
Zhu WH, MacIntyre ANicosia RF . Regulation of angiogenesis by vascular endothelial growth factor and angiopoietin-1 in the rat aorta model: distinct temporal patterns of intracellular signaling correlate with induction of angiogenic sprouting. Am J Pathol 2002; 161(3):823–30.
Asahara T, Chen D, Takahashi T, et al. Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF- induced postnatal neovascularization [see comments]. Circ Res 1998; 83(3):233–40.
Jones MK, Kawanaka H, Baatar D, et al. Gene therapy for gastric ulcers with single local injection of naked DNA encoding VEGF and angiopoietin-1. Gastroenterology 2001; 121(5):1040–7.
Chae JK, Kim I, Lim ST, et al. Coadministration of angiopoietin-1 and vascular endothelial growth factor enhances collateral vascularization. Arterioscler Thromb Vasc Biol 2000; 20(12):2573–8.
Shyu KG, Chang HIsner JM . Synergistic effect of angiopoietin-1 and vascular endothelial growth factor on neoangiogenesis in hypercholesterolemic rabbit model with acute hindlimb ischemia. Life Sci 2003; 73(5):563–79.
Arsic N, Zentilin L, Zacchigna S, et al. Induction of functional neovascularization by combined VEGF and angiopoietin-1 gene transfer using AAV vectors. Mol Ther 2003; 7(4):450–9.
Thurston G, Rudge JS, Ioffe E, et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med 2000; 6(4):460–3.
Gamble JR, Drew J, Trezise L, et al. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ Res 2000; 87(7):603–7.
Kim I, Kim JH, Moon SO, Kwak HJ, Kim NGKoh GY . Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Oncogene 2000; 19(39):4549–52.
Teichert-Kuliszewska K, Maisonpierre PC, Jones N, et al. Biological action of angiopoietin-2 in a fibrin matrix model of angiogenesis is associated with activation of Tie2. Cardiovasc. Res. 2001; 49:659–70.
Mochizuki Y, Nakamura T, Kanetake HKanda S . Angiopoietin 2 stimulates migration and tube-like structure formation of murine brain capillary endothelial cells through c-Fes and c-Fyn. J Cell Sci 2002; 115(Pt–1):175–83.
Gale NW, Thurston G, Hackett SF, et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell 2002; 3(3):411–23.
Goede V, Schmidt T, Kimmina S, Kozian DAugustin HG . Analysis of blood vessel maturation processes during cyclic ovarian angiogenesis. Lab Invest 1998; 78(11):1385–94.
Lobov IB, Brooks PCLang RA . Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci USA 2002; 99(17):11205–10.
Dumont DJ, Gradwohl G, Fong GH, et al. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev 1994; 8(16):1897–909.
Sato TN, Tozawa Y, Deutsch U, et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel for- mation. Nature 1995; 376(6535):70–4.
Lindahl P, Johansson BR, Leveen PBetsholtz C . Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997; 277(5323):242–5.
Tian S, Hayes AJ, Metheny-Barlow LJ Li LY . Stabilization of breast cancer xenograft tumour neovasculature by angiopoietin-1. Br J Cancer 2002; 86(4):645–51.
Iurlaro M, Scatena M, Zhu WH, Fogel E, Wieting SLNicosia RF . Rat aorta-derived mural precursor cells express the Tie2 receptor and respond directly to stimulation by angiopoietins. J Cell Sci 2003; 116(Pt 17)(Pt–17):3635–43.
Park YS, Kim NHJo I . Hypoxia and vascular endothelial growth factor acutely up-regulate angiopoietin-1 and Tie2 mRNA in bovine retinal pericytes. Microvasc Res 2003; 65(2):125–31.
Tian S, Metheny-Barlow, L.J., Hayes, A . and Li, L.-Y . Direct action of Angiopoietin-1 on mesenchymal cells: Stabilization of tumor neovasculature may contribute to tumor growth inhibition. Proc Am Asso Cancer Res 2002; 43:500.
Stratmann A, Risau WPlate KH . Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am J Pathol 1998; 153(5):1459–66.
Ding H, Roncari L, Wu X, et al. Expression and hypoxic regulation of angiopoietins in human astrocytomas. Neuro-oncol 2001; 3(1):1–10.
Audero E, Cascone I, Zanon I, et al. Expression of angiopoietin-1 in human glioblastomas regulates tumor-induced angiogenesis: in vivo and in vitro studies. Arterioscler Thromb Vasc Biol 2001; 21(4):536–41.
Hatanaka H, Abe Y, Naruke M, et al. Significant correlation between interleukin 10 expression and vascularization through angiopoietin/TIE2 networks in non-small cell lung cancer. Clin Cancer Res 2001; 7(5):1287–92.
Wong MP, Chan SY, Fu KH, et al. The angiopoietins, tie2 and vascular endothelial growth factor are differentially expressed in the transformation of normal lung to non- small cell lung carcinomas. Lung Cancer 2000; 29(1):11–22.
Tanaka S, Mori M, Sakamoto Y, Makuuchi M, Sugimachi KWands JR . Biologic significance of angiopoietin-2 expression in human hepatocellular carcinoma. J Clin Invest 1999; 103(3):341–5.
Chen L, Yang Z, Wang GWang C . Expression of angiopoietin-2 gene and its receptor Tie2 in hepatocellular carcinoma. J Tongji Med Univ 2001; 21(3):228–30, 235.
Moon WS, Rhyu KH, Kang MJ, et al. Overexpression of VEGF and angiopoietin 2: a key to high vascularity of hepatocellular carcinoma? Mod Pathol 2003; 16(6):552–7.
Mitsuhashi N, Shimizu H, Ohtsuka M, et al. Angiopoietins and Tie-2 expression in angiogenesis and proliferation of human hepatocellular carcinoma. Hepatology 2003; 37(5):1105–13.
Etoh T, Inoue H, Tanaka S, Barnard GF, Kitano SMori M . Angiopoietin-2 is related to tumor angiogenesis in gastric carcinoma: possible in vivo regulation via induction of proteases. Cancer Res 2001; 61(5):2145–53.
Brown LF, Dezube BJ, Tognazzi K, Dvorak HFYancopoulos GD . Expression of Tie1, Tie2, and angiopoietins 1, 2, and 4 in Kaposi's sarcoma and cutaneous angiosarcoma. Am J Pathol 2000; 156(6):2179–83.
Eggert A, Ikegaki N, Kwiatkowski J, Zhao H, Brodeur GMHimelstein BP . High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin Cancer Res 2000; 6(5):1900–8.
Bunone G, Vigneri P, Mariani L, et al. Expression of angiogenesis stimulators and inhibitors in human thyroid tumors and correlation with clinical pathological features. Am J Pathol 1999; 155(6):1967–76.
Tanaka F, Ishikawa S, Yanagihara K, et al. Expression of angiopoietins and its clinical significance in non-small cell lung cancer. Cancer Res 2002; 62(23):7124–9.
Sfiligoi C, de Luca A, Cascone I, et al. Angiopoietin-2 expression in breast cancer correlates with lymph node invasion and short survival. Int J Cancer 2003; 103(4):466–74.
Ahmad SA, Liu W, Jung YD, et al. The effects of angiopoietin-1 and -2 on tumor growth and angiogenesis in human colon cancer. Cancer Res 2001; 61(4):1255–9.
Oh H, Takagi H, Suzuma K, Otani A, Matsumura MHonda Y . Hypoxia and vascular endothelial growth factor selectively up-regulate angiopoietin-2 in bovine microvascular endothelial cells. J Biol Chem 1999; 274(22):15732–9.
Zhang L, Yang N, Park JW, et al. Tumor-derived vascular endothelial growth factor up-regulates angiopoietin-2 in host endothelium and destabilizes host vasculature, supporting angiogenesis in ovarian cancer. Cancer Res 2003; 63(12):3403–12.
Peoch M . Farion R, Hiou A, Le Bas JF, Pasquier BRemy C . Immunohistochemical study of VEGF, angiopoietin 2 and their receptors in the neovascularization following microinjection of C6 glioma cells into rat brain. Anticancer Res 2002; 22(4):2147–51.
Vajkoczy P, Farhadi M, Gaumann A, et al. Microtumor growth initiates angiogenic sprouting with simultaneous expression of VEGF, VEGF receptor-2, and angiopoietin-2. J Clin Invest 2002; 109(6):777–85.
Koga K, Todaka T, Morioka M, et al. Expression of angio-poietin-2 in human glioma cells and its role for angiogenesis. Cancer Res 2001; 61(16):6248–54.
Yu QStamenkovic I . Angiopoietin-2 is implicated in the regulation of tumor angiogenesis. Am J Pathol 2001; 158(2):563–70.
Zagzag D, Hooper A, Friedlander DR, et al. In situ expression of angiopoietins in astrocytomas identifies angiopoietin-2 as an early marker of tumor angiogenesis [In Process Citation]. Exp Neurol 1999; 159(2):391–400.
Hawighorst T, Skobe M, Streit M, et al. Activation of the tie2 receptor by angiopoietin-1 enhances tumor vessel maturation and impairs squamous cell carcinoma growth. Am J Pathol 2002; 160(4):1381–92.
Mandriota SJPepper MS . Regulation of angiopoietin-2 mRNA levels in bovine microvascular endothelial cells by cytokines and hypoxia. Circ Res 1998; 83(8):852–9.
Pichiule PLaManna JC . Angiopoietin-2 and rat brain capillary remodeling during adaptation and deadaptation to prolonged mild hypoxia. J Appl Physiol 2002; 93(3):1131–9.
Shim WS, Teh M, Mack POGe R . Inhibition of angiopoietin-1 expression in tumor cells by an antisense RNA approach inhibited xenograft tumor growth in immunodeficient mice. Int J Cancer 2001; 94(1):6–15.
Shim WS, Teh M, Bapna A, et al. Angiopoietin 1 promotes tumor angiogenesis and tumor vessel plasticity of human cervical cancer in mice. Exp Cell Res 2002; 279(2):299–309.
Hayes AJ, Huang WQ, Yu J, et al. Expression and function of angiopoietin-1 in breast cancer. Br J Cancer 2000; 83(9):1154–60.
Currie MJ, Gunningham SP, Han C, et al. Angiopoietin-1 is inversely related to thymidine phosphorylase expression in human breast cancer, indicating a role in vascular remodeling. Clin Cancer Res 2001; 7(4):918–27.
Ahmad SA, Liu W, Jung YD, et al. Differential expression of angiopoietin-1 and angiopoietin-2 in colon carcinoma. A possible mechanism for the initiation of angiogenesis. Cancer 2001; 92(5):1138–43.
Larcher F, Franco M, Bolontrade M, et al. Modulation of the angiogenesis response through Ha-ras control, placenta growth factor, and angiopoietin expression in mouse skin carcinogenesis. Mol Carcinog 2003; 37(2):83–90.
Enholm B, Paavonen K, Ristimaki A, et al. Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene 1997; 14(20):2475–83.
Stoeltzing O, Ahmad SA, Liu W, et al. Angiopoietin-1 inhibits tumour growth and ascites formation in a murine model of peritoneal carcinomatosis. Br J Cancer 2002; 87(10):1182–7.
Stoeltzing O, Ahmad SA, Liu W, et al. Angiopoietin-1 inhibits vascular permeability, angiogenesis, and growth of hepatic colon cancer tumors. Cancer Res 2003; 63(12):3370–7.
Singh M, Holash, J ., Yancopoulos, G.D . and Hanahan, D . Overexpression of Angiopoietin-1 modulates tumor progression in a transgenic mouse model of squamous carcinogenesis. Proc. Am. Asso. Cancer Res. 2002; 43:500.
Kuwabara TCogan DG . Mural cells of the retinal capillaries. Arch Ophthalmol 1963; 69:492–502.
Feldman PS, Shneidman DKaplan C . Ultrastructure of infantile hemangioendothelioma of the liver. Cancer 1978; 42(2):521–7.
Wang Y, Li Y, Zhu G, et al. [Ultrastructural and immunohistochemical characteristics of pericytes during neovascularization in breast carcinoma]. Zhonghua Bing Li Xue Za Zhi 2000; 29(3):176–9.
Egginton S, Hudlicka O, Brown MD, Graciotti LGranata AL . In vivo pericyte-endothelial cell interaction during angiogenesis in adult cardiac and skeletal muscle. Microvasc Res 1996; 51(2):213–28.
Egginton S, Zhou AL, Brown MDHudlicka O . The role of pericytes in controlling angiogenesis in vivo. Adv Exp Med Biol 2000; 476:81–99.
Crocker DJ, Murad TMGeer JC . Role of the pericyte in wound healing. An ultrastructural study. Exp Mol Pathol 1970; 13(1):51–65.
Orlidge AD'Amore PA . Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J Cell Biol 1987; 105(3):1455–62.
Antonelli-Orlidge A, Saunders KB, Smith SRD'Amore PA . An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci USA 1989; 86(12):4544–8.
Sato YRifkin DB . Inhibition of endothelial cell movement by pericytes and smooth muscle cells: activation of a latent transforming growth factor-beta 1-like molecule by plasmin during co-culture. J Cell Biol 1989; 109(1):309–15.
Korff T, Kimmina S, Martiny-Baron GAugustin HG . Blood vessel maturation in a 3-dimensional spheroidal coculture model: direct contact with smooth muscle cells regulates endothelial cell quiescence and abrogates VEGF responsiveness. Faseb J 2001; 15(2):447–57.
Lafleur MA, Forsyth PA, Atkinson SJ, Murphy GEdwards DR . Perivascular cells regulate endothelial membrane type-1 matrix metalloproteinase activity. Biochem Biophys Res Commun 2001; 282(2):463–73.
Osada H, Tokunaga T, Hatanaka H, et al. Gene expression of angiogenesis related factors in glioma. Int J Oncol 2001; 18(2:):305–9.
Hata K, Udagawa J, Fujiwaki R, Nakayama K, Otani HMiyazaki K . Expression of angiopoietin-1, angiopoietin-2, and Tie2 genes in normal ovary with corpus luteum and in ovarian cancer. Oncology 2002; 62(4):340–8.
Martoglio AM, Tom BD, Starkey M, Corps AN, Charnock-Jones DSSmith SK . Changes in tumorigenesis- and angiogenesis-related gene transcript abundance profiles in ovarian cancer detected by tailored high density cDNA arrays. Mol Med 2000; 6(9):750–65.
Wurmbach JH, Hammerer P, Sevinc S, Huland HErgun S . The expression of angiopoietins and their receptor Tie-2 in human prostate carcinoma. Anticancer Res 2000; 20(6D):5217–20.
Yoshida Y, Oshika Y, Fukushima Y, et al. Expression of angiostatic factors in colorectal cancer. Int J Oncol 1999; 15(6):1221–5.
Currie MJ, Gunningham SP, Turner K, et al. Expression of the angiopoietins and their receptor Tie2 in human renal clear cell carcinomas; regulation by the von Hippel-Lindau gene and hypoxia. J Pathol 2002; 198(4):502–10.
Nagata J, Kijima H, Hatanaka H, et al. Angiopoietin-1 and vascular endothelial growth factor expression in human esophageal cancer. Int J Mol Med 2002; 10(4):423–6.
Ijland SA, Jager MJ, Heijdra BM, Westphal JRPeek R . Expression of angiogenic and immunosuppressive factors by uveal melanoma cell lines. Melanoma Res 1999; 9(5):445–50.
Mitsutake N, Namba H, Takahara K, et al. Tie-2 and angiopoietin-1 expression in human thyroid tumors. Thyroid 2002; 12(2):95–9.
Giuliani N, Colla S, Lazzaretti M, et al. Proangiogenic properties of human myeloma cells: production of angiopoietin-1 and its potential relationship to myeloma-induced angiogenesis. Blood 2003; 102(2):638–45.
Uneda S, Matsuno F, Sonoki T, Tniguchi I, Kawano FHata H . Expressions of vascular endothelial growth factor and angiopoietin-2 in myeloma cells. Haematologica 2003; 88(1):113–5.
Takahama M, Tsutsumi M, Tsujiuchi T, et al. Enhanced expression of Tie2, its ligand angiopoietin-1, vascular endothelial growth factor, and CD31 in human non-small cell lung carcinomas. Clin Cancer Res 1999; 5(9):2506–10.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
METHENY-BARLOW, L., LI, L. The enigmatic role of angiopoietin-1 in tumor angiogenesis. Cell Res 13, 309–317 (2003). https://doi.org/10.1038/sj.cr.7290176
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7290176
Keywords
This article is cited by
-
The dynamic roles of the bladder tumour microenvironment
Nature Reviews Urology (2022)
-
Extracellular vesicles derived from Wharton’s Jelly mesenchymal stem cells inhibit the tumor environment via the miR-125b/HIF1α signaling pathway
Scientific Reports (2022)
-
Identification of significant genes as prognostic markers and potential tumor suppressors in lung adenocarcinoma via bioinformatical analysis
BMC Cancer (2021)
-
Candidate protein biomarkers in pancreatic neuroendocrine neoplasms grade 3
Scientific Reports (2020)
-
Effects of thiamine and fenofibrate on high glucose and hypoxia-induced damage in cell models of the inner blood-retinal barrier
Acta Diabetologica (2020)
