
Abstract
Purpose Clinical assessment of outcome of corneal replacement with a synthetic cornea, AlphaCor™, in patients considered at too high risk for conventional penetrating keratoplasty with donor tissue to be successful, but excluding indications such as end-stage dry eye that might be suited to traditional prosthokeratoplasty.
Methods All patients in the multicentre clinical trial were managed according to an approved protocol, with Ethics Committee approval in each centre. Preoperative visual acuity ranged from perception of light (PL) to 6/60 (20/200). Implantation was by means of an intralamellar technique, with a conjunctival flap in most cases. Tissues anterior to the optic were removed as a secondary procedure.
Results Up to 30 November 2001, 40 AlphaCor™ devices had been implanted in 38 patients, of mean age 60 years. Follow-up ranged from 0.5 months to 3 years. There had been one extrusion (2.5%) and four cases (10%) where a device had been removed due to melt-related complications. All five of these cases received a donor corneal graft after the device was removed, with these grafts remaining anatomically satisfactory and epithelialised to date. Corneal melts in AlphaCor™ recipients were found to be strongly associated with a history of ocular herpes simplex infection. Two further devices (5%) were removed owing to reduced optic clarity after presumed drug-related deposition, and have been successfully replaced with second devices. Mean preoperative best-corrected visual acuity was hand movements. Visual acuities after surgery ranged from PL to 6/6−2 (20/20−2).
Conclusions Early results suggest that the AlphaCor™, previously known as the Chirila keratoprosthesis (Chirila KPro), has a low incidence of the complications traditionally associated with keratoprostheses and can be effective in restoring vision in patients considered untreatable by conventional corneal transplantation. Importantly, the device can be replaced with a donor graft in the event of development of a significant complication. A history of ocular herpes simplex is a contraindication to AlphaCor™ implantation. Ongoing monitoring of clinical outcomes in all patients will allow the indications for AlphaCor™, as opposed to donor grafts, to be determined.
Similar content being viewed by others

Introduction
AlphaCor™, previously known as the Chirila KPro, is a synthetic corneal replacement that was developed in response to a clinical need for an alternative to human donor tissue in cases where a high risk of failure precludes penetrating keratoplasty with donor tissue (abbreviated here as dPK). A prospective clinical study was undertaken to assess the outcomes of AlphaCor™ surgery (‘synthetic’ penetrating keratoplasty, sPK) in patients considered at too high risk for dPK. Outcomes were considered in terms of complications and acuity outcomes.
Materials and methods
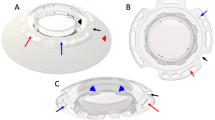
The multicentre clinical investigation was sponsored by the Lions Eye Institute of Western Australia and the devices were supplied by Argus Biomedical Pty Ltd. The AlphaCor™ artificial corneas are made entirely from the hydrophilic polymer poly(2-hydroxyethyl methacrylate), or PHEMA, and comprise a central optic region surrounded by a concentric skirt that is chemically identical yet higher in water content (Figure 1).1 The peripheral skirt is macroporous and allows biointegration by tissue ingrowth.2 The optic and the surrounding skirt are fused together by means of an interpenetrating polymer network that prevents dehiscence, leakage or downgrowth at this interface.3

AphaCor™ keratoprosthesis.
Patients were recruited for the clinical investigation if they were adults whose corneal pathology was unsuited to management by means of a conventional corneal graft owing to the presence of risk factors for failure. Preoperative visual acuities were in the range hand movements (HM) to 6/60 (20/200). Eyes were required to be without active inflammation and with controlled intraocular pressure prior to surgery, and eyes with less than 50% normal tear production by Schirmer's test were excluded. Patients were fully informed of the possible benefits and risks prior to recruitment, and Ethics Committee approval was obtained in each clinical investigation centre.
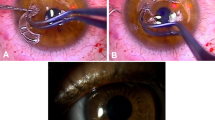
Devices were implanted by means of a two-stage procedure.4 During the first stage, a 360° conjunctival peritomy was made and the corneal epithelium was debrided. A superior 180°, half-thickness incision was made in the sclera 1 mm posterior to the limbus and this incision was then extended using a lamellar dissecting blade to form a flap of the superior cornea at 50% depth. The blade was then used to continue into the inferior cornea to create an intralamellar pocket of 3.5 mm radius (Figure 2). Retraction of the superior flap inferiorly allowed the centre of the posterior lamella to be visualised and trephined with a 3-mm disposable trephine, entering the anterior chamber. The AlphaCor™ was then placed within the pocket with its centre overlying the posterior lamellar opening. The superior lamellar flap was then replaced and sutured at the limbus with interrupted 10/0 nylon (Figure 3). A Gunderson conjunctival flap was then created and brought over the corneal surface.

Creation of lamellar pocket to receive AlphaCor™.

Insertion of AlphaCor™ within the pocket after a 3-mm trephination of the lamella posterior to the device optic.
After 12 weeks, the second stage of the procedure was performed. The tissues anterior to the AlphaCor™ optic (conjunctiva and anterior corneal lamella) were removed under topical anaesthetic using a 3-mm trephine and scissors (Figure 4). Following this procedure, vision was assessed and clinical care continued according to a protocol. Postoperative care included field tests, disc examination using a 90D lens, and estimation of intraocular pressure using Pulsair™ (Keeler Instruments, Inc., PA, USA) air tonometers, TonoPen™ (Automated Ophthalmics, Inc., MD, USA) electronic tonometers, or the Proview™ (Bausch & Lomb, Inc., NY, USA) phosphene tonometer. Postoperative medication comprised guttae prednisolone 0.5% and chloramphenicol 0.3% for the initial postoperative period, along with topical tetracycline 1% ointment (Latycin®, Biochemie GmbH, Vienna, Austria), which was continued four times daily as the only long-term medication. The first 10 patients in the series additionally received guttae medroxyprogesterone 1%, but this was subsequently stopped to conform to regulatory requirements (FDA, USA), as the drug is not approved for use with a keratoprosthesis. Conformance with this medication regime was encouraged, but it was acknowledged that some patients occasionally required different or additional medication, such as for pre-existing glaucoma. All medication prescribed was recorded.

Appearance of AlphaCor™ in situ after the second stage of surgery: optic exposed as a full-thickness corneal replacement.
Results
Of the patients recruited, 82% were male and 18% were female, perhaps partly because of the relatively high incidence of cases with a history of work-related trauma or chemical injury as the original pathology. The age of patients ranged from 20 to 83 years, mean 60 years.
The total number of AlphaCor™ devices implanted to the end of November 2001 was 40, with follow-up ranging from 2 weeks to 36 months (mean 10.1 months). A total of 26 cases (65%) had completed stage II surgery (opening of the tissues anterior to the optic) prior to 30 November 2001. A total of 12 cases (30%) had achieved more than 12 months follow-up, and 24 cases (60%) had achieved more than 6 months follow-up. The device had been retained to this date in 33 cases (82.5%), removed and replaced with donor tissue in five cases (12.5%, including one case classified as an extrusion, where the device had been largely separated from the cornea by a thick retroprosthetic downgrowth), and removed and replaced with a second AlphaCor™ in two cases (5%) (Figure 5).

(a) AlphaCor™ implant status at 30 November 2001. (b) Kaplan–Meier curve of device retention up to 30 November 2001.
Dystrophies (22%), chemical injuries (20%) and herpes simplex (20%) were the commonest indications for the primary corneal replacement (donor graft) in the patients recruited to the study. Most patients in the series had two or more failed donor corneal grafts prior to recruitment to the study, and the mean number of prior grafts was 1.6 (Figure 6). A total of 15% of cases had not received a donor corneal graft owing to a judgement that failure would be inevitable. Where there had been a prior donor graft, AlphaCor™ surgery was not performed within 12 months of the most recent graft to minimise risks of dehiscence at the host–graft interface during implantation. Where there had been one or more prior donor corneal grafts, the previous graft had remained clear for a mean period of 1.5 years, range 0 days to 9 years.

AlphaCor™ cases (% series) with a history of 0–4 prior failed donor corneal grafts.
In all, 40% of patients had four quadrants of deep corneal vessels prior to implantation (Figure 7), with the mean number of quadrants of corneal vascularisation being 2.3.

AlphaCor™ cases (% series) with 0–4 quadrants of deep corneal vascularisation.
Five cases had a cataractous lens and required concurrent AlphaCor™ implantation, cataract extraction, and IOL placement. The remainder were phakic with a clear lens (30.8%), pseudophakic with a posterior chamber intraocular lens (IOL) (43.6%), pseudophakic with a Kelman-style anterior chamber IOL (7.7%) or with another type of anterior chamber or iris fixated IOL (5.2%), or were aphakic (12.8%) both before and after implantation.
Outcome data for every patient at every visit were collected for analysis, with no case lost to follow-up. No patient has lost vision so as to be unable to perceive light. Preoperative best-corrected visual acuity (VA) ranged from perception light (PL) to 6/60 (20/200), mean HM. At 30 November 2001, the current best-corrected visual acuity ranged from PL to 6/24 (20/80), mean count fingers (CF). The visual acuities postimplantation varied between visits depending upon the diameter of anterior flap opening (which constricted in some patients, limiting vision until reopened) and refractive correction, with acuities up to 6/6−2 (20/20−2) being recorded during follow-up in some cases. Devices produced at an early stage of the trial tended to produce a myopic outcome, such that the best acuities were achieved with a plano-rigid gas-permeable contact lens. However, concerns about the risk of contamination resulted in lens wear being discontinued, and the resulting limitation in acuity in some patients as a result of this has reduced the mean improvement in VA on current recording. Concurrent pathologies diagnosed prior to AlphaCor™ implantation that limited visual potential were present in 20 (50%) cases, predominantly glaucoma (27%), diabetic retinopathy (10%), and age-related macular degeneration (5%). Preoperative and best postoperative acuities (uncorrected and best-corrected) during follow-up are shown in Figure 8.

Change in minimum, mean, and maximum VA for AlphaCor™ recipients: best-corrected VA pre-AlphaCor™ implantation; best unaided VA recorded after AlphaCor™ surgery, and best-corrected VA recorded after AlphaCor™ surgery. VA is scored in lines of visual acuity from 0=no perception light, 1=perception light, 2=hand movements, 3=count fingers, 4=6/60 (20/200), 5=6/36 (20/120), 6=6/24 (20/80), 7=6/18 (20/60), 8=6/12 (20/40), 9=6/9 (20/30), and 10=6/6 (20/20).
Slightly over half of the patients (52.5%) have remained free to date of any complication requiring treatment. Complications can be categorised as anatomical complications related to loss of corneal stromal tissue, optic complications related to deposition or surface damage and device unrelated complications, as summarised in Table 1.
The commonest category of complication was that related to stromal melting adjacent or anterior to the device skirt. This was observed in 30% of cases and was strongly associated with a history of ocular herpes simplex. Of eight cases with a history of ocular herpes simplex infection, six (75%) have experienced a melt and three of these have required device explantation and replacement with donor tissue. This compares with an 18.8% incidence of stromal melts in patients with no known history of herpes simplex virus (HSV). Four cases of stromal melting developed in patients who had had cataract removal concurrent with, or after, AlphaCor™ implantation, where soft lens matter may have been retained, promoting chronic ocular inflammation. No cases of rhegmatogenous retinal detachment or of endophthalmitis were reported.
The other main category of complication relates to surface spoilation or of deposition of substances within the hydrogel optic such that vision is reduced. There have been two cases (5%) of mild–moderate surface spoilation with the appearance of a contact lens ‘jelly-bump’ type. These deposits require regular cleaning. However, the majority of cases have had no surface deposition and there have been no cases of giant papillary conjunctivitis.
In 15% of cases, there has been some intragel optic deposition. Two cases (5% of series) were noted to have developed a brownish discoloration. Both of these patients were heavy smokers, one smoking filterless cigarettes, and the other additionally received topical levobunolol (Betagan®, Allergan, Inc., CA, USA). Both smoking and Betagan® have subsequently been found by us (unpublished data) to cause a brown deposition in vitro (Figure 9). A diffuse, whitish deposition was observed in three cases (7.5%), in each case starting as a mild, superficial deposition early after stage II surgery, and progressing to a denser whiteness, severely affecting vision. In all these cases, medications other than chloramphenicol and Latycin® had been used prior to the onset of the deposition. One of the affected devices has been explanted for analysis to date and demonstrated the presence of calcium phosphate within the deposits (Figure 10). It is tentatively suggested that initial adsorption of certain medications or combinations of medications might have provided initiation sites for subsequent calcium deposition. In one further case (2.5%), following a period of contact lens wear, two small, greyish branching lesions were observed within the peripheral part of the device optic. All cultures were negative. These lesions responded to topical amphotericin and broad-spectrum antimicrobials, but the toxicity of these drugs caused some stromal melting over the adjacent area of device skirt and required repair with a lamella of donor corneal tissue. Subsequently, greyish fluffy balls were seen in the plane between the device optic and the lamellar graft (Figure 11). This was presumed to be a recurrence of fungal infection and the device was explanted for testing and replaced with a new device.

(a) Optic deposition presumed related to topical levobunolol. (b) AlphaCor™ soaked in guttae levobunolol in vitro.

(a) Onset of a diffuse grey-white deposition within the optic. (b) Explanted specimen showing that the white deposition stains with Alizarin Red.

(a) Appearance of grey-white intragel lesions at 3 o'clock, which responded to a combination of topical antifungal and antibiotic medication. (b) Appearance of recurrent, presumed fungal, lesions between the device optic and the overlying lamellar tissue.
Discussion
Early data are encouraging in that most cases maintain an anatomically stable eye with an improved visual acuity. Patients report that eyes are comfortable, and appreciate the relatively ‘low-maintenance’ regimen and absence of requirement for systemic steroids and other cytotoxic drugs. Cosmesis, although not normal, is acceptable (Figure 12).

Appearance of AlphaCor™ in a patient 24 months after device implantation.
The incidence of the severe complications traditionally associated with keratoprostheses appears to be low, in part because of the relatively noninvasive implantation procedure (ie lack of requirement for lens or iris removal, vitrectomy or muscle detachment, and avoidance of encroaching upon the drainage angle). Further, in most cases it is possible to remove a device in the event of complications and replace it with a second device or with donor tissue in the same manner that a failed graft is replaced. No patient to date has lost an eye or developed endophthalmitis. This contrasts with several other published keratoprosthesis series where high incidences of serious complications are noted within the first 6 months.5 The flexibility of the hydrogel optic allows useful estimations of intraocular pressure to be made, and disc examinations and field tests can be performed after surgery.
Complications were found to fall into two categories: anatomical or melt-related, and optic deposition/spoilation. There was a statistically significant association between both melt-related complications and device removals and a history of ocular HSV infection, such that a history of ocular HSV is now considered a contraindication for AlphaCor™ surgery.6
The most significant types of optic deposition were (1) a brown discoloration, attributed to topical medication (levobunolol) and smoking, and (2) a white deposition whose natural history is not fully understood but is thought to be related to topical drug deposition that may, in some cases, allow secondary calcium deposition within the altered gel. Research into risk factors and mechanisms of depositions within hydrogels continues so as to minimise the incidence of this complication through avoidance of precipitating factors. Contact lens wear may be a risk factor for the development of infective lesions and is not currently recommended for AlphaCor™ recipients.
Despite limitations imposed by both pre-existing pathology in 50% of cases and the suboptimal quality of the optic surfaces of AlphaCor™ devices supplied during the early part of the clinical investigation, a mean improvement in visual acuity was recorded and maintained throughout the study period. The AlphaCor™ artificial cornea appears able to allow restoration of a patient's full visual potential.
These outcomes have provided feedback to surgeons using the device, with protocols being altered to reflect newly recognised risk factors. Data collection is a central part of the AlphaCor™ study and a detailed database continues to be compiled so that data of the expanding clinical series can be reanalysed as the follow-up period increases.
References
Chirila TV, Constable IJ, Crawford GJ, Russo AV . Method of producing a keratoprosthesis. US patent 5,458,819, 17 October 1995.
Vijayasekaran S, Hicks CR, Chirila TV, Fitton JH, Clayton AB, Lou X et al. Histologic evaluation during healing of hydrogel core-and-skirt keratoprostheses in the rabbit eye. Cornea 1997; 16: 352–359.
Chirila TV, Vijayasekaran S, Horne R, Chen YC, Dalton P, Constable IJ et al. Interpenetrating polymer network (IPN) as a permanent joint between the elements of a new type of artificial cornea. J Biomed Mater Res 1994; 28: 745–753.
Crawford GJ, Hicks CR, Lou X, Vijayasekaran S, Tan D, Mulholland B et al. The Chirila keratoprosthesis: phase I human clinical trial. Ophthalmology 2002; 109: 883–889.
Hicks CR, Fitton JH, Chirila TV, Crawford GJ, Constable IJ . Keratoprostheses: advancing toward a true artificial cornea. Surv Ophthalmol 1997; 42: 175–189.
Hicks CR, Crawford GJ, Tan DT, Snibson GR, Sutton GL, Gondhowiardjo TD et al. Outcomes of implantation of an artificial cornea AlphaCor: the effects of prior ocular herpes simplex infection. Cornea 2002; in press.
Acknowledgements
We thank Susan Davies for assistance with the preparation of the manuscript.
Lions Eye Institute owns the patent rights to the AlphaCor™ device. CR Hicks, GJ Crawford, X Lou, TV Chirila, and IJ Constable have financial interests in Argus Biomedical Pty Ltd, a company that is licensed to manufacture and distribute the AlphaCor™ device.
Supported by a grant from the National Health and Medical Research Council of Australia.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was presented in part at the 27th Annual Meeting of the Castroviejo Cornea Society, New Orleans, November 2001
Rights and permissions
About this article
Cite this article
Hicks, C., Crawford, G., Lou, X. et al. Corneal replacement using a synthetic hydrogel cornea, AlphaCor™: device, preliminary outcomes and complications. Eye 17, 385–392 (2003). https://doi.org/10.1038/sj.eye.6700333
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6700333
