
Abstract
Aims To assess the clinical features, pathology, mortality (systemic outcome) and ocular complications (visual outcome) of a cohort of patients treated for intraocular lymphoma.
Methods Retrospective case analysis of medical records and review of pathology of a consecutive series of patients presenting with intraocular lymphoma in Melbourne over 11 years between 1990 and 2000. Categorical factors influencing survival were examined by the Kaplan–Meier estimator and groups compared with the log rank test.
Results A total of 14 patients were included. The median age of onset of symptoms was 62.5 years. Most were male (64%) and had bilateral eye involvement (64%). The commonest presentation was vitritis in 12 patients, with a median delay of 4 months before diagnosis. In all, 10 patients had B-cell lymphoma, three patients T-cell lymphoma and one null-cell. Four patients had prior systemic lymphoma. Eight patients had primary central nervous system non-Hodgkin's lymphoma (PCNSL). Treatment included combined radiation to the eye and chemotherapy in 10 patients. Complications of radiotherapy included cataract in five (50%), dry eyes in four (40%), punctate keratopathy in two (20%), radiation retinopathy in two (20%), and optic atrophy in one (10%). A total of 11 patients died of lymphoma (79%). One has residual ocular disease, while two have survived for more than 5 years from initial presentation. Although currently disease free, one of these has a poor visual outcome with acuity less than 6/60 secondary to ocular complications of treatment.
Conclusions Our study had 29% with prior systemic lymphoma, 57% associated with PCNSL and 14% with intraocular disease only. Overall survival is low (21%) and relapses common in those surviving beyond 12 months. Visual outcome in survivors is very poor due, in large part, to significant complications from radiotherapy.
Similar content being viewed by others

Introduction
Intraocular lymphoma, originally named ‘ocular reticulum cell sarcoma’, is an uncommon condition, first described in 1951.1 Disease may be either primary or secondary, but is more commonly associated with primary central nervous system non-Hodgkin's lymphoma (PCNSL), defined as lymphoma limited to the craniospinal axis without systemic disease.2 PCNSL is a multifocal process and may involve the brain parenchyma, meninges, intradural spinal cord, and the eye. Primary intraocular lymphoma is itself considered a subset of PCNSL, and is defined as infiltration by malignant lymphoid cells of the uveal tract, retina, vitreous or optic nerve head, in the absence of systemic lymphoma, and may occur with or without central nervous system (CNS) disease.
The incidence of PCNSL has increased over the past few decades to 6.6%3 of primary intracranial tumours in some neuropathological series3,4,5 in both immunocompromised and immunocompetent patients. This is only in part because of the HIV-1 epidemic, as there also appears to be an increasing incidence of sporadic, non-HIV-related disease. It is likely that this is associated with an increased incidence of intraocular lymphoma, although no data are as yet available.
The ideal treatment of intraocular lymphoma is difficult to determine because of the rarity of the disease. Combined chemoradiation regimens have most commonly been utilised, although for PCNSL there has been investigation of combination chemotherapy with less or no radiotherapy as primary treatment, aiming to reduce neurotoxicity, especially in elderly patients.6,7 Improved survival for intraocular lymphoma has been attributed to heightened awareness of the entity among ophthalmologists with earlier diagnosis and instigation of treatment. However, overall the prognosis of the disease remains poor. There are little data specifically on long-term sequelae of treatment for intraocular lymphoma in the literature, and we undertook this study to investigate survival and morbidity.
Patients, materials and methods
A total of 14 patients were identified from the ocular immunology clinic at the Royal Victorian Eye and Ear Hospital, from the private practices of the immunology clinic consultants and also from the records of the Anatomical Pathology department at St Vincent's Hospital, all in Melbourne, Australia, and diagnosed between 1990 and 2000. Clinical data, including patient's age at the onset of disease, gender, medical history, clinical findings, laboratory investigations, mode of diagnosis, interval between the onset of symptoms and final diagnosis, type of therapy, complications of therapy, disease course and duration of survival, and final ocular outcome, were obtained from various hospital and treatment centre records. One patient still alive 10 years after initial diagnosis of intraocular lymphoma, with very poor acuity bilaterally, was excluded from the patient sample. He was diagnosed with sarcoidosis on mediastinal biopsy 7 years after initial ocular presentation, and his original vitreous specimen was unavailable for re-examination.
Pathology
Histology and cytology specimens from St Vincent's Anatomical Pathology laboratory and four other pathology laboratories were reviewed by one of the authors (PMcK). The lymphoma cases were classified by the reviewing pathologist according to the Revised European and American Lymphoma (REAL) classification. Samples included cytology of nine vitrectomy specimens, four cerebrospinal fluid (CSF) samples, and two anterior chamber taps. Biopsies included two of brain, two lymph nodes, one sclera and iris tissue, one retinal biopsy, one extradural lumbar mass, and one enucleated globe. Cytospin preparations were made from vitreous fluid, some of which were stained with Giemsa and Papanicolaou for diagnosis. Others were air-dried for phenotyping by immunocytochemistry for antibodies including B-cell antigen CD20 and T-cell antigen CD3 (antibodies from DAKO). Flow cytometry was performed in patient 5 on a CSF sample.
Statistical analysis
Data were analysed with Stata version 7.1 (STATA Corporation, College Station, TX, USA). Results were expressed as actual numbers or percentage for categorical variables. Categorical factors influencing survival were examined by the Kaplan–Meier estimator and groups compared with the log rank test.8 Primary intraocular lymphoma and secondary disease were considered separately in examining factors including lymphoid phenotype. The effect of age was assessed by Cox regression.9 A probability <0.05 was considered statistically significant.
Results
Demographic data
Of 14 patients, nine were male (64%). The median age of onset of symptoms was 62.5 years with range from 24 to 79 years (Table 1). All the patients were immunocompetent and all were HIV negative. Nine patients (64%) had bilateral disease.
Anatomical distribution and clinical features
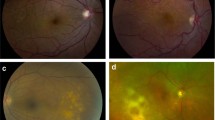
The ocular features at presentation were varied with several signs/symptoms seen concurrently in many patients (Table 2) (Figure 1 and Figure 2). The commonest symptoms reported by patients were blurred vision in 12 patients (86%) and floaters in six patients (43%). In our series, vitritis was the commonest presenting feature in 12 patients (86%). Seven patients (50%) had choroidal involvement (six of these had multifocal choroiditis, defined as well-demarcated subretinal pale or pigmented areas). B-scan ultrasound in patient 8 showed diffuse choroidal and scleral involvement.

Clinical photograph (patient 6) demonstrating evidence of vasculitic change in superotemporal quadrant and large confluent choroidal infiltrates.

Angiogram (patient 1) demonstrating vasculitis and disc leakage.
The four patients with secondary lymphoma (prior systemic lymphoma) showed anterior uveitis in all, bilateral disease in 50%, choroidal involvement, vitritis and disc swelling in 75%, increased intraocular pressure in 50%, but none showed retinitis. By contrast, those patients with primary ocular lymphoma +/− CNS disease showed anterior uveitis in 30%, bilateral disease in 70%, vitritis in 90%, retinitis in 20%, choroidal involvement in 40%, optic disc swelling in 10%, but none showed raised intraocular pressure.
Comparing the ocular features in the three patients with T-cell lymphoma (cases 1, 7, 8) with 10 patients with B-cell disease showed approximately similar proportions with bilateral disease, anterior uveitis, vitritis, and choroidal involvement. However, T-cell cases showed a higher proportion with disc swelling. No T-cell lymphoma patients had raised intraocular pressure.
Diagnosis
Median time to diagnosis after the initial ocular presentation was 4 months, (range 0–58 months). Diagnosis of lymphoma was made most commonly from vitreous fluid in six out of 14 patients (43%). In the other eight patients, the diagnosis was by a variety of other means including CSF cytology in two, brain biopsy in two, spinal extradural tissue biopsy in one, anterior chamber tap in one, sclera and iris biopsies in one, and lymph node biopsy in one.
Four patients had secondary lymphoma with systemic (non-CNS) lymphoma diagnosed between 2 and 23 months prior to the intraocular involvement (Table 1). This included three B-cell lymphomas (follicular, mantle cell and diffuse large B-cell) and one peripheral T-cell lymphoma.
In the three patients with T-cell lymphoma, there were five negative cytology or biopsy specimens before diagnosis. These included two vitreous fluids, two CSF samples and one retinal biopsy. Patient 7 had negative CSF, vitreous and retinal biopsies before a brain biopsy revealed T-cell lymphoma.
Pathology
Pathological diagnosis included nine diffuse large B cell (64.5%), one large cell with immunocytochemistry negative for B and T markers (7%), one mantle cell lymphoma (7%), and three T-cell lymphomas (21.5 %). The null-cell case was from an outside institution where flow cytometry was not performed and no samples for immunocytochemistry were prepared, but the cells did not resemble plasmacytoma. The T-cell lymphomas included one natural killer (NK/T-cell) lymphoma presenting with ocular-CNS disease and ensuing rapidly fatal systemic disease (flow cytometry CD2+, CD3−, CD4−, CD7+, CD8−, CD16−, CD56+), one rare primary T-cell CNS lymphoma with ocular involvement, and one peripheral T-cell lymphoma.
Extraocular involvement
Four patients (29%) had prior systemic lymphoma (three B cell, one T cell) present for 2–23 months before ocular diagnosis (Table 1). One patient (NK/T-cell lymphoma) developed systemic disease 2 months after concurrent eye and CNS disease. Two of the patients with prior systemic disease (both B-cell lymphomas) also developed CNS disease, one 6 months prior to the eye and the other 19 months after.
Eight patients (57%) had primary CNS lymphoma, which included five B cell, two T cell, and one null-cell lymphoma. One patient developed intraocular B-cell lymphoma 8 months after PCNSL was diagnosed. Four patients developed concurrent eye and CNS disease (two B cell, two T cell) and three patients (two B cell, one null-cell) developed CNS disease at 10, 24, and 45 months after the diagnosis of intraocular lymphoma. Only one of eight (12.5%) patients with PCNSL developed systemic disease and pathology in that case was the high-grade NK/T-cell lymphoma with rapid demise within 10 weeks of diagnosis.
Treatment
A total of 10 patients (71%) received both chemotherapy and radiation treatment to the eye, including eight bilateral (Table 1 and 3). Treatment regimens varied between institutions and included CHOP (cyclophosphamide, doxorubicin/adriamycin, vincristine, prednisolone), methotrexate (both intravenous and intrathecal), dexamethasone, cytosine arabinoside and cisplatin, and EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide and doxorubicin). One patient with ongoing systemic disease received five cycles of chemotherapy and autologous stem-cell transplant. Only one patient received intravitreal methotrexate for recurrent ocular disease, with poor clinical response. Four patients (29%) had chemotherapy treatment alone and one patient had a unilateral enucleation for recurrent ocular disease after having initial chemoradiation.
A wide range of radiotherapy doses was administered to 10 patients (Table 3). The most common dose administered was 40 G administered in 20–25 fractions (equivalent to 1.6–2.0 G per fraction).
Prognosis and clinical follow-up (Table 1)
Two patients with prior systemic lymphoma suffered systemic and ocular relapses 19 and 34 months following initial remission, which responded to further chemotherapy in one patient, but was followed by further systemic relapses and ultimately death in the other. Two patients with PCNSL suffered intraocular relapse of lymphoma at 18 and 28 months, which responded to further chemotherapy, but recurred again in one, resulting in enucleation, as the patient declined intravitreal chemotherapy. The other patient suffered a fatal CNS relapse at 48 months and died.
In total, 11 patients (79%) died as a result of lymphoma. The range of time from intraocular diagnosis until death was 1–52 months (median 9 months). The median overall survival in all patients from diagnosis was 16 months. Patients with no CNS involvement had improved survival with two of four (50%) alive in contrast to only one of 10 patients (10%) with CNS lymphoma (eight primary, two secondary). A similar proportion of patients with secondary and primary ocular lymphoma died of lymphoma (75 and 80%, respectively).
Three patients (21%) are still alive. One patient with large B-cell lymphoma has residual unilateral ocular disease with visual acuity 6/36 in both eyes, but no CNS involvement at 24 months. Two (one peripheral T cell, one large B cell) have survived for more than 5 years since ocular diagnosis (101 and 103 months) with both currently in remission. However, one is blind with visual acuity less than 6/60 bilaterally secondary to radiation retinopathy and optic atrophy (Figure 3). This patient has also had excisional biopsy for carcinoma in situ of the corneal epithelium, 62 months following radiotherapy. The other long-term survivor without disease has only one eye after enucleation of the right eye for recurrent intraocular lymphoma. Visual acuity in the left eye is 6/18 and he is receiving topical antiglaucoma medication.

Clinical photograph (patient 1) showing gross retinal ischaemia and optic atrophy secondary to radiation retinopathy. Argon laser photocoagulation scars evident.
Statistical analysis
By log rank test, none of the variables examined were associated with a statistical difference in survival. Factors thought to influence survival (presence of systemic lymphoma, primary CNS lymphoma, lymphoma phenotype, age) were not significant (P>0.05, log rank test and Cox regression analysis).
Complications of radiotherapy
A variety of complications were seen in seven patients (70%) receiving radiotherapy (Table 3). The most common complication was cataract severe enough to necessitate surgery in five (50%) patients. Other commonly encountered complications were keratoconjunctivitis sicca in four (40%), punctate keratopathy in two (20%), radiation retinopathy in two (20%), and optic atrophy in one (10%). Rubeotic glaucoma and carcinoma in situ of the limbus were each encountered in one patient.
Discussion
This study confirmed the frequent (64%) bilateral occurrence of intraocular lymphoma and similar demographics of disease, mainly in an elderly male population.10,11,12,13,14 Our youngest patient was an immunocompetent 24-year-old woman with NK/T-cell disease whose case has already been reported.15 The most common clinical presentation in our study was vitritis (86%). The ocular features at presentation varied in patients with primary ocular lymphoma +/− CNS involvement and patients with secondary disease.
Intraocular lymphoma is a subset of PCNSL, of which 98% are B-cell lineage and usually large high-grade tumours.16 However, 21% of patients in our series had T-cell lymphoma, similar to the figure of 21% T-cell type found by Brown et al17 in their large review of 57 published cases of intraocular lymphoma.
T-cell intraocular lymphomas are rare compared with B-cell lymphomas. Of 23 published cases with clinical data,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35 nine (39%) were associated with mycosis fungoides (primary T-cell malignancy of skin),20,24,25,27,29,30,31,32,34,35 10 (43%) with systemic (nonmycosis) lymphoma,17,18,19,22,23,26,27,28,33 two (9%) with CNS and intraocular lymphoma only,17,20 and two (9%) were restricted to the eye only.18,21 Therefore, systemic disease is found in 82% of patients with T-cell intraocular lymphoma. Those patients with underlying mycosis fungoides usually have a long history of cutaneous lymphoma ranging from 2 to 30 years (median 7 years) and die within weeks to months of developing intraocular lymphoma.24,25,31,32,34 Of those with systemic (nonmycosis) T-cell lymphoma, two-thirds of reported patients have died with a median survival of 8 months after diagnosis of the eye disease.22,23,27,28,33 However, two patients with documented systemic T-cell lymphoma are still alive with long follow-up of 60 and 95 months.17,18 The two reported cases of intraocular T-cell lymphoma without systemic or CNS lymphoma were alive with a relatively short follow-up of 6 and 18 months.18,21 Follow-up of those two cases was available only for one of the T-cell PCNSL involving the eye, who was alive at 27 months,17 but the other HIV-positive patient has presumably died.20 Interestingly, there is a male predominance of 2:1 for T-cell intraocular lymphoma, and the median age of onset for men is 46 years compared to 59 years for women. Primary T-cell CNS lymphoma only accounts for 1.5–3.5% of all PCNSL and occurs in a younger, mainly male population who are usually immunocompetent.36,37,38
Although intraocular lymphoma is most commonly associated with PCSNL, 29% of our patients had secondary lymphoma. Two of these developed secondary CNS lymphoma in the course of their illness, one prior to and one after diagnosis of ocular lymphoma. In all, 57% of our patients had PCNSL and then presented concurrently with the ocular disease in half of the patients, after ocular lymphoma in 37.5% and in only one, preceding ocular disease. Only one (12.5%) of these eight patients with PCNSL developed systemic disease, comparable to the incidence of less than 10% systemic disease previously reported.21,38 Two of our patients (14%) had ocular lymphoma with no proven CNS or systemic disease, although one patient developed a paraneoplastic cerebellar syndrome. However, autopsy was not performed.
Diagnosis of intraocular lymphoma is commonly difficult.11,12 We observed a median delay of 4 months after onset of symptoms in our patients, and the mean delay (10.2 months) was similar to or shorter than that reported by other groups.10,11,13,39 Large-cell lymphoma, which is usually of the B-cell phenotype, may be recognised on either air-dried or fixed cytospin preparations of CSF or vitreous, which have been handled correctly (Figure 4A). However, difficulties in cytological diagnosis are well recognised because the large lymphoma cells may be fragile and susceptible to damage during collection; there may be a large mixed inflammatory and necrotic infiltrate, and viable malignant cells may be scarce.11 Difficulties were encountered in the diagnosis of two cases of T-cell lymphoma because of the variation in size of the neoplastic lymphocytes, ranging from small to large, mimicking a reactive process (Figure 4B). In our series, all of the five false negative cytological and biopsy specimens occurred in patients with T-cell lymphoma, and all had received prior corticosteroid therapy. We now try and avoid performing biopsies in patients on high doses of corticosteroid therapy as it is recognised that some lymphomas will initially respond to such treatment, steroids being cytotoxic to PCNSL cells.11

(A) (Upper plate) – Vitreous specimen (patient 6) demonstrating numerous large atypical lymphoid cells (B-cell). Pap stain. Original magnification × 200. (B) (Lower plate) – Brain biopsy (patient 7) showing infiltrates of atypical lymphoid cells ranging from small to large. Immunocytochemistry for T-cell marker CD3. Original magnification × 200.
Treatment regimens in our series involved chemoradiation (71%) and chemotherapy only (29%). Although PCNSL is highly radiosensitive and radiotherapy can produce complete remission, disease relapses (ocular or CNS) are common and cure is rare, with median survival generally 12–18 months only.40 Chemotherapy may be administered by intrathecal, intravenous, or intravitreal routes. The only patient in our series treated with intravitreal methotrexate for recurrent disease died shortly thereafter. Ocular and systemic relapse after initial response was seen in two of our patients with systemic lymphoma and two patients with PCNSL also suffered intraocular relapse. All recurrences were noted 18 months or more after initial ocular diagnosis and occurred in over half of those surviving over 12 months. Mortality in our study was high (79%) with a median survival of only 16 months after diagnosis. Factors including presence of systemic or primary CNS lymphoma, lymphoma phenotype, and age had no statistically significant impact on survival. Two patients survived longer than 5 years (14%), significantly better than the 5-year survival of less than 5% reported by Merchant and Foster.41
Complications as a result of radiotherapy were common, affecting 70% of the patients. The most frequent was cataract in 50%. However, these patients had other precipitants for cataract development including previous ocular surgery, steroid treatment, chronic posterior uveitis, and the generally older age of the population. Dry eye was also relatively common (40%), while the more serious complications of radiation retinopathy and optic atrophy were noted in 20 and 10%, respectively. Since radiation retinopathy typically develops 18–36 months after treatment,42 and 62.5% of patients receiving radiotherapy who died in our series did so within 18 months, it is likely that many patients died before complications such as radiation retinopathy could manifest. One survivor has poor visual acuity less than 6/60 secondary to radiation retinopathy and optic atrophy (Figure 3). The patient excluded from the study also has acuity less than 6/60 bilaterally secondary to radiation retinopathy.
The possibility of radiotherapy complications, especially that of radiation retinopathy in long-term survivors, creates significant treatment dilemmas for intraocular lymphoma management. Patients who have good vision and mild ocular involvement may be better off without radiotherapy after consideration of potential complications and life expectancy. Adequate management of the patient's dry eyes, cataracts, and laser photocoagulation of radiation retinopathy is important.
The use of intravitreal methotrexate appears to offer good local control of the disease.43,44,45,46 This has been used in patients as initial therapy and for recurrent disease. The treatment protocols incorporating induction and maintenance phases are relatively complex and prolonged. There are a number of reported incidences of visual deterioration associated with this treatment, although the relationship with the methotrexate is still uncertain.46 Also if intravitreal treatment fails, there is a potential for spread of lymphoma from the eye.
It is clear that there are serious problems with the current protocols of chemoradiation. If irradiation is utilised, follow-up is important with early management of any complications, such as cataract and dry eyes. Although most intraocular lymphoma is radiosensitive, in general, disease treated with radiotherapy alone will relapse. Those protocols using no radiation and advocating intravitreal methotrexate for primary treatment may be preferable, especially for potentially long-term survivors. However, it is difficult to select such patients and the treatment is not without difficulties. Further studies on ocular outcomes with both protocols are necessary.
References
Cooper EL, Riker JL . Malignant lymphoma of the uveal tract. Am J Ophthalmol 1951; 34: 1153–1158.
Fine HA, Mayer RJ . Primary central nervous system lymphoma. Ann Intern Med 1993; 119: 1093–1104.
Miller DC, Hochberg FH, Harris NL, Gruber ML, Louis DN, Cohen H . Pathology with clinical correlations of primary central nervous system non-Hodgkin's lymphoma. The Massachusetts General Hospital experience 1958–1989. Cancer 1994; 74: 1383–1397.
Eby NL, Grufferman S, Flannelly CM, Schold SC Jr, Vogel FS, Burger PC . Increasing incidence of primary brain lymphoma in the US. Cancer 1988; 62: 2461–2465.
Corn BW, Marcus SM, Topham A, Hauck W, Curran WJ Jr. Will primary central nervous system lymphoma be the most frequent brain tumour diagnosed in the year 2000? Cancer 1997; 79: 2409–2413.
Freilich RJ, Delattre JY, Monjour A, DeAngelis LM . Chemotherapy without radiation therapy as initial treatment for primary CNS lymphoma in older patients. Neurology 1996; 46: 435–439.
Sandor V, Stark-Vancs V, Pearson D, Nussenblat R, Whitcup SM, Brouwers P et al. Phase II trial of chemotherapy alone for primary CNS and intraocular lymphoma. J Clin Oncol 1998; 16: 3000–3006.
Kaplan EL, Meier P . Non-parametric evaluation from incomplete observation. J Am Stat Assoc 1958; 53: 457–481.
Cox DR . Regression models and life tables. J R Stat Soc 1972; 34: 187–220.
Freeman LN, Schachat AP, Knox DL, Michels RG, Green WR . Clinical features, laboratory investigations, and survival in ocular reticulum cell sarcoma. Ophthalmology 1987; 94: 1631–1638.
Whitcup SM, de Smet MD, Rubin BI, Palestine AG, Martin DF, Burnier M Jr et al. Intraocular lymphoma. Clinical and histopathological diagnosis. Ophthalmology 1993; 100: 1399–1406.
Peterson K, Gordon KB, Heinemann MH, DeAngelis LM . The clinical spectrum of ocular lymphoma. Cancer 1993; 72: 843–849.
Cassoux N, Merle-Beral H, Leblond V, Bodaghi B, Miléa D, Gerber S et al. Ocular and central nervous system lymphoma: clinical features and diagnosis. Ocul Immunol Inflamm 2000; 8; 243–250.
Char DH, Ljung BM, Miller T, Phillips T . Primary intraocular lymphoma (ocular reticulum cell sarcoma) diagnosis and management. Ophthalmology 1988; 95: 625–630.
Hunyor AP, Harper A, O'Day J, McKelvie PA . Ocular-central nervous system lymphoma mimicking posterior scleritis with exudative retinal detachment. Ophthalmology 2000; 107: 1955–1959.
Kleihues P, Cavenee WK (eds). Malignant lymphomas. In: WHO Classification of Tumours. Pathology and Genetics of Tumours of the Nervous System. IARC Press: Lyon, 2000; pp. 198–203.
Brown SM, Jampol LM, Cantrill HL . Intraocular lymphoma presenting as retinal vasculitis. Surv Ophthalmol 1994; 39: 133–140.
Char DH, Ljung BM, Deschênes J, Miller TR . Intraocular lymphoma: immunological and cytological analysis. Br J Ophthalmol 1988; 72: 905–911.
Goldey SH, Stern GA, Oblon DJ, Mendenhall NP, Smith LJ, Duque RE . Immunophenotypic characterization of an unusual T-cell lymphoma presenting as anterior uveitis. A clinicopathological case report. Arch Ophthalmol 1989; 107: 1349–1353.
Davis JL, Viciana AL, Ruiz P . Diagnosis of intraocular lymphoma by flow cytometry. Am J Ophthalmol 1997; 124: 362–372.
Ridley ME, McDonald HR, Sternberg Jr P, Blumenkranz MS, Zarbin MA, Schachat AP . Retinal manifestations of ocular lymphoma (reticulum cell sarcoma). Ophthalmology 1992; 99: 1153–1160.
Saga T, Ohno S, Matsuda H, Ogasawara M, Kikuchi K . Ocular involvement by a peripheral T-cell lymphoma. Arch Ophthalmol 1984; 102: 399–402.
Kohno T, Uchida H, Inomata H, Fukushima S, Takeshita M, Kikuchi M . Ocular manifestations of adult T-cell leukaemia/lymphoma. A clinicopathologic study. Ophthalmology 1993; 100: 1794–1799.
Williams GC, Holz E, Lee AG, Font RL . T-cell lymphoproliferative disorder of vitreous associated with mycosis fungoides. Arch Ophthalmol 2000; 118: 278–280.
Lois N, Hiscott PS, Nash J, Wong D . Immunophenotypic shift in a case of mycosis fungoides with vitreous invasion. Arch Ophthalmol 2000; 118: 1692–1694.
Yahalom C, Cohen Y, Averbukh E, Anteby I, Amir G, Pe'er J . Bilateral iridociliary T-cell lymphoma. Arch Ophthalmol 2002; 120: 204–207.
Goeminne JC, Brouillard A, Jaumain P, Ferrant A, Snyers B, De Potter P . Bilateral granulomatous panuveitis as initial presentation of diffuse systemic T-cell lymphoma. Ophthalmologica 1999; 213: 323–326.
Jensen OA, Kiss K, Johansen S . Intraocular T-cell lymphoma mimicking a ring melanoma. First manifestation of systemic disease. Graefes Arch Clin Exp Ophthalmol 1994; 232: 148–152.
Foerster HC . Mycosis fungoides with intraocular involvement. Trans Am Acad Ophthalmol Otol 1960; 64: 308–313.
Gärtner J . Mycosis fungoides mit Beteiligung der Aderhaut. Klin Monatsblat Augenheilkd 1989; 131: 61–69.
Keltner JL, Fritsch E, Cykiert RC, Albert DM . Mycosis fungoides. Intraocular and central nervous system involvement. Arch Ophthalmol 1977; 95: 645–650.
Wolter JR, Leenhouts TM, Hendrix RC . Corneal involvement in mycosis fungoides. Am J Ophthalmol 1963; 55: 315–322.
Reim H, Dieler R, Wessing A . Non-Hodgkin-Lymphom mit dem Erscheinungsbild einer Chorioretinitis. Fortschr Ophthalmol 1990; 87: 557–559.
Erny BC, Egbert PR, Peat IM, Shorrock K, Rosenthal AR . Intraocular involvement with subretinal pigment epithelium infiltrates by mycosis fungoides. Br J Ophthalmol 1991; 75: 698–701.
Leitch RJ, Rennie IG, Parsons MA . Ocular involvement in mycosis fungoides. Br J Ophthalmol 1993; 77: 126–127.
Bednar MM, Salerni A, Flanagan ME, Pendlebury WW . Primary central nervous system T-cell lymphoma. J Neurosurg 1991; 74: 668–672.
Grant JW, Isaacson PG . Primary central nervous system lymphoma. Brain Pathology 1992; 2: 97–109.
McCue MP, Sandrock AW, Lee JM, Harris NL, Hedley-Whyte ET . Primary T-cell lymphoma of the brainstem. Neurology 1993; 43: 377–381.
Rothova A, Ooijman F, Kerkhoff F et al. Uveitis masquerade syndromes. Ophthalmology 2001; 108: 386–399.
DeAngelis LM . Current management of primary central nervous system lymphoma. Oncology 1995; 9: 63–78.
Merchant A, Foster CS . Primary intraocular lymphoma. Int Ophthalmol Clin 1997; 37: 101–115.
Parsons JT, Bova FJ, Mendenhall WM, Million RR, Fitzgerald CR . Response of the normal eye to high dose radiotherapy. Oncology 1996; 10: 837–847.
Fishburne BC, Wilson DJ, Rosenbaum JT, Neuwelt EA . Intravitreal methotrexate as an adjunctive treatment of intraocular lymphoma. Arch Ophthalmol 1997; 115: 1152–1156.
de Smet MD, Vancs VS, Kohler D, Solomon D, Chan CC . Intravitreal chemotherapy for the treatment of recurrent intraocular lymphoma. Br J Ophthalmol 1999; 83:448–551.
de Smet MD . Management of non-Hodgkin's lymphoma with intravitreal methotrexate. Bull Soc Belge Ophthalmol 2001; 279: 91–95.
Smith JR, Rosenbaum JT, Wilson DJ, Doolittle ND, Siegal T, Neuwelt EA et al. Role of intravitreal methotrexate in the management of primary central nervous system lymphoma with ocular involvement. Ophthalmology. 2002; 109: 1709–1716.
Acknowledgements
We would like to thank Dr KH Liew for reading and reviewing the discussion as related to radiation oncology. We also thank the numerous ophthalmologists, general practitioners, neurologists and radiation oncologists, as well as the multitude of medical record staff for their assistance and allowing access to clinical data. The staff of the various pathology labs (Alfred Hospital, Cabrini Hospital, Monash Medical Centre and Dorevitch Pathology) are acknowledged for allowing access and review of slides where available.
Author information
Authors and Affiliations
Corresponding author
Additional information
None of the authors have any proprietary interest related to this article. This work was presented in part in poster format at the Association of Research in Vision and Ophthalmology Annual Meeting, Fort Lauderdale, Florida (May 2002)
Rights and permissions
About this article
Cite this article
Hoffman, P., McKelvie, P., Hall, A. et al. Intraocular lymphoma: a series of 14 patients with clinicopathological features and treatment outcomes. Eye 17, 513–521 (2003). https://doi.org/10.1038/sj.eye.6700378
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6700378
Keywords
This article is cited by
-
Epidemiology and survival outcomes of patients with primary intraocular lymphoma: a population-based analysis
BMC Ophthalmology (2022)
-
A proposed protocol of intravitreal injection of methotrexate for treatment of primary vitreoretinal lymphoma
Eye (2022)
-
An audit of retinal lymphoma treatment at the University of California San Francisco
Eye (2020)
