
Abstract
Aims To report a Bangladeshi family displaying intrafamilial phenotypic heterogeneity of lattice corneal dystrophy type I (LCDI) and to identify the causative mutation.
Methods Molecular genetic analysis was performed on DNA extracted from all members of the family. Exons of BIGH3gene were amplified by polymerase chain reaction. Gene mutation and polymorphisms were identified by heteroduplex and sequence analyses. Segregation of the mutation in the family was confirmed by restriction digestion of amplified gene fragments.
Results A heterozygous C → T transition at the first nucleotide position of codon 124 of the BIGH3gene was detected in the three affected members and not in the unaffected members of the family.
Conclusions This is the first report of BIGH3gene mutation in a Bangladeshi family with phenotypic heterogeneity. This study confirms that BIGH3gene screening should be undertaken for proper classification of corneal dystrophy, especially in the absence of histopathological examination.
Similar content being viewed by others
Introduction
Corneal dystrophies are a genetically and clinically heterogeneous group of disorders that occurs most frequently bilaterally and are manifested around the time of puberty, progressing slowly throughout life.1 Most corneal dystrophies are inherited as an autosomal dominant trait.2
Several phenotypic variants of corneal dystrophies with an autosomal dominant inheritance were reported to result from specific mutation(s) in BIGH3 (or transforming growth factor, β-induced [TGFBI]) gene on chromosome 5q31 (Table 1). Lattice corneal dystrophy type I (LCDI, OMIM 122200),3, 4, 5, 6 LCD type IIIA (LCDIIIA),7, 8, 9, 10, 11 intermediate type I/IIIA (LCDI/IIIA),9, 10, 11, 12 deep stromal LCD (LCDIV),13, 14, 15 granular corneal dystrophy (GCD, OMIM 121900),3, 4, 15 Avellino corneal dystrophy (ACD, OMIM 121900),3, 4, 16 Thiel Behnke corneal dystrophy or corneal dystrophy of Bowman layer type 2 (CDB2, OMIM 602082)3, 4, 17, 20, and Reis-Bücklers' corneal dystrophy (RBCD, OMIM 121900) all result from mutations in BIGH3.17, 18, 19, 20 These conditions are defined by their clinical manifestations and the histological characteristics of corneal deposits.21
LCDI (OMIM 122200) has an autosomal dominant mode of inheritance with variable expressivity. The disease presents towards the end of the first or second decade of life and slowly progresses to cause discomfort and visual impairment, usually before the sixth decade. It is characterised by thin greyish, linear branching deposits of amyloid material that progressively accumulate in the subepithelial and stromal layers of the cornea.22 The specific cause of the amyloid deposits is unclear, however, they may be secondary to collagen degeneration or produced by abnormal keratocytes.
Histopathologically, the white dots and lattice lines consist of amyloid in the corneal stroma, which is confirmed histochemically by the pink to orange staining of the deposits by Congo red.2
It has been reported that both R124C and L518P mutations were responsible for LCDI in both Caucasian and Japanese families.3, 4, 5, 6
In this study, a Bangladeshi family has been studied both clinically and genetically. Our results revealed a heterozygous C to T transition at the first nucleotide position of codon 124 in the BIGH3 gene in all affected members. Clinically, the phenotype of LCDI in the proband and his brother was not typical. However, the finding of only single mutation in the three affected members of the family, including the mother with her typical LCDI phenotype, further indicates that screening of BIGH3 gene is a mandatory step to arrive at an unambiguous diagnosis of corneal dystrophy.
Materials and methods
Report of cases
The cases were examined by MFE and LAF.
-
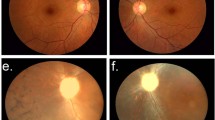
Case II-2 (the proband): A 10-year-old boy who was referred to Moorfields Eye Hospital complaining of intermittent ocular irritation and mild visual impairment. The episodes of ocular irritation began at the age of 5 years. The patient's best corrected visual acuity was 6/18 in both eyes. His intraocular pressure (IOP) was 12 mmHg in the right eye and 11 mmHg in the left eye. He had no history of keratoplasty.Slit-lamp biomicroscopic examination revealed bilateral confluent dense geographic subepithelial corneal opacities resembling that of RBCD, but with intervening very fine lattice lines (Figure 1a).
Figure 1 
Slit lamp biomicroscopic findings: (a) the right eye of the proband (II-2) showing geographic subepithelial deposits (atypical LCDI), (b) the right eye of the proband's brother (II-3) showing diffuse superficial corneal haze with superimposed amorphous deposits (atypical LCDI) (c) the left eye of the proband's mother (I-1) showing the lattice lines typical of LCDI.
-
Case II-3 (the proband's brother): An 8-year-old boy affected by the same phenotypic presentation as the proband and having no history of eye surgery (Figure 1b).
-
Case I-1 (the proband's mother): A 42-year-old female had also been examined. She had a past history of photophobia, lacrimation, and chronic ocular irritation since childhood. Her visual acuity was 6/24 in both eyes and her IOP was 14 mmHg in the right eye and 12 mmHg in the left eye. She had no history of keratoplasty. Slit-lamp examination showed dense subepithelial opacities with fine lattice lines in the superficial and midstromal layers of both corneas (Figure 1c). A full medical history was obtained and no systemic manifestations of amyloidosis such as cranial and peripheral neuropathies, skin changes such as lichen amyloidosis, cutis laxa, blepharochalasis, protruding lips, and mask facies were detected.
-
Case I-2 (the proband's father): Unaffected as determined by the slit-lamp biomicroscopic examination.
-
Case II-1 (the proband's brother): Unaffected as determined by the slit-lamp biomicroscopy.

Methods
The study had the approval of Moorfields Eye hospital Ethics Committee and conformed to the tenets of the Declaration of Helsinki. An informed consent from all participants was obtained and blood samples were collected from the parents, the two affected, and the one unaffected offspring.
Genomic DNA was extracted from peripheral leucocytes by standard methods and was amplified using the polymerase chain reaction (PCR). The intronic primers used are those listed in (Table 2) spanning exon/intron boundaries. Amplification was performed in a DNA thermal cycler using the following conditions: 3 min at 94°C followed by 35 cycles of 94°C for 1 min, 60–64°C for 1 min, and 72°C for 1 min, with a final extension step at 72°C for 5 min.
DNA from the participants was amplified using the intronic primers (4–16) of the BIGH3 gene and analysed on a heteroduplex gel. The amplified DNA for the exons that showed positive results with heteroduplex analysis was purified using a Qiaquick PCR purification kit (Qiagen) and sequenced using an automatic fluorescent DNA sequencer (ABI Prism 373A, Perkin Elmer, Foster City, CA, USA) following the manufacturer's instructions. Nucleotide sequences for the coding regions were compared with the nucleotide sequence of the published BIGH3 human cDNA.23 Following identification of the C → T mutation by sequencing, a cosegregation study was performed using AvaII restriction enzyme (Promega). The recognition site for AvaII is 5′…G▾G(A/T)CC…3′ and the C → T change abolishes the recognition site for the mutated allele. The novel sequence alteration found in intron 7 was tested in 100 control chromosomes using DdeI restriction enzyme (Promega) with recognition sequence 5′…C▾TNAG…3′. The T → C change abolishes the recognition site of the enzyme.
Results
Mutation screening of BIGH3 gene by heteroduplex analysis revealed positive results for exons 4, 7, 11, and 12 in all affected members with the exception of exon 7, which was positive for the mother only. No changes were detected in other exons. Direct sequencing analysis of the proband (II-2) and his mother (I-1) for exon 4 revealed a heterozygous base pair transition at nucleotide position 417 (C → T), which converts an arginine at codon 124 into cysteine as shown in Figure 2. This mutation has previously been reported in LCDI.3, 4, 5, 6 The unaffected members of the family did not have the mutation as determined by a cosegregation study of the restriction digest analysis in which the mutation abolishes the recognition site of AvaII. The three affected members showed one undigested product of the expected size (310 bp) for the mutant allele plus two restriction digest products (190 bp and 110 bp) from the normal allele. Unaffected members depicted two restriction digest fragments (190 bp and 110 bp) as shown in Figure 3. Sequence analysis of the same exon revealed a polymorphism in intron 4 (IVS4-16C → T) that was previously reported.4, 12

Direct sequencing of exon 4 of the BIGH3 gene. (a) the proband (II-2) showing heterozygous single base pair transition (CGC → TGC, R124C). (b) the unaffected father (I-2) showing the wild-type allele.

The results of cosegregation study by restriction analysis. M=Øx174-HaeIII marker; lane 1: undigested 310 bp PCR product; lanes 2 and 5: AvaII digested product from unaffected members (II-1) and (I-2) respectively, showing the digested wild-type alleles (190 bp and 110 bp); lanes 3, 4, and 6: AvaII digested product from affected members (I-1), (II-2), (II-3), respectively, carrying the R124C mutation showing the undigested mutated allele (310 bp); lane 7 contains no sample.
Sequence analysis of the other three PCR products that were aberrantly migrating as identified by heteroduplex analysis showed polymorphisms that did not change the amino-acid sequence of the protein. The polymorphisms were identified in intron 7 (IVS7+46T → C), exon 11 at nucleotide 1416 with C → T change and in exon 12 at nucleotide 1620 with T → C change.
Two of these polymorphisms have previously been identified,4, 12 however, the polymorphism in intron 7 is novel. Although the polymorphism in intron 7 was detected in the mother affected by the typical picture of LCDI and was not detected in the proband and his brother, it did not cosegregate with the disease and was detected in a control population of the same ethnic origin at a frequency of 92 out of 100 chromosomes.
Discussion
In this study, we report a family from Bangladesh with two different phenotypes, the two affected offspring have a phenotype that resemble that of RBCD, whereas the mother has a typical LCDI phenotype.
All affected members of the family have an R124C mutation in the BIGH3 gene. The same mutation was found in Caucasian3 and Japanese patients with LCDI.24 This suggests that this mutation occurs independently in several ethnic groups.5
Molecular genetic analysis of the BIGH3 gene in the present family did not reveal the R555Q mutation previously found in Caucasians with CDB23 or the R124L mutation found in Japanese individuals with RBCD.17 Also, it did not reveal the L518P previously reported with LCDI.6 Instead, the same R124C mutation previously associated with LCDI was found in both the mother (LCDI) and the two affected offspring (atypical LCDI).
It has been postulated that RBCD may be a variant of LCD, with a mild to nonexistent deep stromal pathology.25 Our findings based on clinical and genetic studies of the family support this theory. It is possible that the mother's phenotype originally resembled that of her offspring and that the development of lattice lines in her cornea is secondary to chronic irritation produced by numerous episodes of superficial erosions.
Another possible explanation for the observed variant phenotype is the effect of a modifier locus in which other genes affect the phenotypic expression of the mutated BIGH3 gene. There are well-documented examples of intrafamilial variable expressivity for deafness in humans, which have been attributed to modifier gene.26
The fact that the affected members in this family had not undergone keratoplasty made the categorisation of corneal dystrophy difficult. However, the genetic analysis declared that all affected members in this family belong to one type of corneal dystrophy.
Thus, we conclude that the possibility of RBCD being a variant of LCDI is reinforced by the study of this family and that BIGH3 gene screening should be mandatory for proper classification of corneal dystrophies, especially in the absence of histopathological examination. Also, the detection of R124C mutation in a Bangladeshi family for the first time adds to the previous reports that BIGH3 mutations occur independently in different ethnic origins.
References
McTigue JW . The human cornea: a light and electron microscopic study of the normal cornea and its alterations in various dystrophies. Trans Am Ophthalmol Soc 1967; 65: 591.
Waring III GO, Mbekeani JN . Corneal dystrophies. In: Leibowitz HW Waring III GO (eds) Corneal disorders. Clinical diagnosis and management, 2nd ed. WB Saunders: Philadelphia, 1998; 245–251.
Munier FL, Korvatska E, Djemai A, Le Paslier D, Zografos L, Pescia G et al. Kerato-epithelin mutations in four 5q31 linked corneal dystrophies. Nat Genet 1997; 15: 247–251.
Korvatska E, Munier FL, Djemai A, Wang MX, Frueh B, Chiou AGY et al. Mutation hot spots in 5q31 linked corneal dysrophies. Am J Hum Genet 1998; 62: 320–324.
Gupta SK, Hodge WG, Damji KF, Guernsey DL, Nuemann PE . Lattice corneal dystrophy type I in a Canadian Kindred is associated with the Arg124 → Cys mutation in the kerato-epithelin gene. Am J Ophthalmol 1998; 125: 547–549.
Endo S, Ha NT, Fujiki K, Hotta Y, Nakayasu K, Yamaguchi T et al. Leu518Pro mutation of the Big-h3 gene causes lattice corneal dystrophy type I. Am J Ophthalmol 1999; 128: 104–106.
Yamamoto S, Okada M, Tsujikawa M, Shimomura Y, Nishida K, Inoue Y et al. A kerato-epithelin (Big-h3) mutation in lattice corneal dystrophy type IIIA (letter). Am J Hum Genet 1998; 62: 719–722.
Kawasaki S, Nishida K, Quantock AJ, Dota A, Bennett K, Kinoshita S . Amyloid and Pro501Thr- mutated ßßßig-h3 gene product colocalize in lattice corneal dystrophy type IIIA. Am J Ophthalmol 1999; 127: 456–458.
Stewart H, Black GC, Donnai D, Bonshek RE, McCarthy J, Morgan S et al. A mutation within exon 14 of the TGFBI (BIGH3) gene on chromosome 5q31 causes an asymmetric, late onset form of lattice corneal dystrophy. Ophthalmology 1999; 106: 964–970.
Dighiero P, Durnat S, Ellies P, D'Hermies, F, Savoldelli, M, Legeais, JM et al. A new mutation (A546T) of the ßig-h3 gene responsible for a French lattice corneal dystrophy type IIIA. Am J Ophthalmol 2000; 129(2): 248–251.
Munier FL, Frueh BE, Othenin Girard P, Uffers S, Cousin P, Wang MX et al. BIGH3 mutation spectrum in corneal spectrum. Inv Ophthalmol Vis Sci 2002; 43: 949–954.
Schmitt-Bernard CF, Guittard C, Arnaud B, Demaille J, Argiles A, Claustres M et al. Big-H3 exon 14 mutations lead to intermediate type I/IIIA of lattice corneal dystrophies. Invest Ophthalmol Vis Sci 2000; 41: 1302–1308.
Fujiki K, Hotta Y, Nakayasu K, Yokoyama T, Tkano T, Yamaguchi et al. A new L527R mutation of the Big-h3 gene in patients with lattice corneal dystrophy with deep stromal opacities. Hum Genet 1998; 103: 286–289.
Hirano K, Hotta Y, Nakamura M, Fujiki K, Kanai A, Yamamoto N . Late onset form of lattice corneal dystrophy caused by Leu527Arg mutation of the TGFBI gene. Cornea 2001; 20: 525–529.
Stewart HS, Ridgway AE, Dixon MG, Bonshek R, Parveen R, Black G . Heterogeneity in granular corneal dystrophy: Identification of three causative mutations in the TGFBI (BIGH3) gene: lesson for corneal amyloidogenesis. Hum Mutat 1999; 14: 126–132.
Fujiki K, Hotta Y, Nakayasu K, Kanai A . Homozygotic patient with Bigh3 gene mutation in granular dystrophy. Cornea 1998; 17: 288–290.
Okada M, Yamamoto S, Tsujikawa M, Watanabe H, Inoue Y, Maeda N et al. Two distinct kerato-epithelin mutations in Reis Bucklers' corneal dystrophy. Am J Ophthalmol 1998; 126: 535–542.
Mashima Y, Nakamura Y, Noda K, Konishi M, Yamada M, Kudoh J et al. A novel mutation of codon 124 (R124L) in the Big H3 gene is associated with a superficial variant of granular corneal dystrophy. Arch Ophthalmol 1999; 117: 90–93.
Rozzo C, Forsarello M, Galleri G, Sole G, Serru A, Orzalesi N et al. A common Big-H3 gene mutation (ΔF540) in a large cohort of Sardinian Reis–Bucklers' corneal dystrophy patients. Hum Mutat 1998; 12: 215–216.
Ridgway AE, Akhtar S, Munier FL, Schorderet DF, Stewart H, Perveen R et al. Ultrastructural and molecular analysis of Bowman's layer corneal dystrophies: an epithelial origin? Invest Ophthalmol Vis Sci. 2000; 41(11): 3286–3292.
Klintworth JK, Valnickova Z, Enghild JJ . Accumulation of Big-h3 gene product in cornea with granular dystrophy. Am J Pathol 1998; 152: 743–748.
Durand L, Resal R, Burillon C . Focus on an antomoclinical entity: Biber–Haab–Dimmer lattice dystrophy. J Fr Ophthalmol 1985; 8: 729.
Skonier J, Naubauer M, Madisen L, Bennett K, Plowman JD, Purchoi AF . cDNA cloning and sequence analysis of Big- H3, a novel gene induced in a human adenocarcinoma cell line after treatment with human transforming growth factor ß. DNA Cell Biol 1992; 11: 511–522.
Mashima Y, Imamura Y, Konishi M, Nagasawa A, Yamada M, Oguchi Y et al. Homogeneity of kerato-epithelin codon 124 mutations in Japanese patients with either of two types of corneal srtromal dystrophy. Am J Hum Genet 1997; 61: 1448–1450.
King RG, Geeraets WJ . Lattice or Reis–Bücklers' corneal dystrophy: a question of stromal pathology. Southern Med. J. 1969; 62: 1163–1169.
Friedman TB, Battey J, Kachar B, Riazuddin S, Noben-Trauth K, Griffith A et al. Modifiers genes of hereditary hearing loss. Curr Opin Neurobiol 2000; 10: 487–493.
Acknowledgements
We thank the members of the family who participated in this study. A research grant from, Egypt Tanta University Hospital, Egypt supported this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
El-Ashry, M., Abd El-Aziz, M., Ficker, L. et al. BIGH3 mutation in a Bangladeshi family with a variable phenotype of LCDI. Eye 18, 723–728 (2004). https://doi.org/10.1038/sj.eye.6701313
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6701313
