





Abstract
Motivation: Storing, annotating and analyzing variants from next-generation sequencing projects can be difficult due to the availability of a wide array of data formats, tools and annotation sources, as well as the sheer size of the data files. Useful tools, including the GATK, ANNOVAR and BEDTools can be integrated into custom pipelines for annotating and analyzing sequence variants. However, building flexible pipelines that support the tracking of variants alongside their samples, while enabling updated annotation and reanalyses, is not a simple task.
Results: We have developed variant tools, a flexible annotation and analysis toolset that greatly simplifies the storage, annotation and filtering of variants and the analysis of the underlying samples. variant tools can be used to manage and analyze genetic variants obtained from sequence alignments, and the command-line driven toolset could be used as a foundation for building more sophisticated analytical methods.
Availability and implementation: variant tools consists of two command-line driven programs vtools and vtools_report. It is freely available at http://varianttools.sourceforge.net, distributed under a GPL license.
Contact: bpeng@mdanderson.org
1 INTRODUCTION
Tracking samples and predicted variants from next-generation sequencing projects often requires building custom analysis pipelines. Data standards such as the Browser Extensible Data (BED) (Hinrichs et al., 2006), General Feature Format and Variant Call Format (VCF) (Danecek et al., 2011) file specifications can be used to represent these variants in a common format, simplifying integration of tools and the construction of these analysis pipelines. Difficulties include the integration of diverse annotation sources and the management of many large intermediate files containing millions of predicted variants and millions more associated annotations for each sample. These annotation sources and intermediate files often have fundamental inconsistencies using either 0- or 1-based coordinates and potentially different genomic builds, which can complicate their management and integration.
For biologists or analysts who have familiarity with programming and running tools from the command line, there are many useful tools that can be integrated into custom pipelines to annotate and filter variants. These tools include ANNOVAR (Wang et al., 2010) and BEDTools (Quinlan and Hall, 2010). However, building effective pipelines that relate variants to their samples and sample attributes (such as cases and controls), while applying multiple annotation sources require a large customization effort. A framework for building pipelines that facilitate simple, reproducible and recurrent analyses is currently lacking. Therefore, we have developed variant tools, a flexible, open-source toolset upon which custom pipelines can be easily constructed. This toolset facilitates the storage of variants (alongside their sample details) as well as the annotation, filtering and reporting of these variants at multiple levels—starting with variant reports based on individual samples to project-wide variant reports.
2 METHODS
variant tools is a command-line driven toolset written in the Python scripting language that incorporates either SQLite or MySQL as a backend database management system. The toolset is used to create a variant project, which is conceptually designed around a master variant table that often consists of millions of variants for all of the samples in a sequencing project along with variant attributes (called fields in variant tools). Variant fields can include sample statistics, which variant tools can generate, or information provided by annotation data sources. Regardless of the source of these fields, they can be used to select, output and analyze genetic variation from the project. As illustrated in Figure 1, analyzing genetic variants from next-generation sequencing projects typically involves four steps, namely importing, annotating, filtering and reporting. variant tools installs easily and sets up a working environment with human genome annotation sources that can be downloaded automatically. Since variant tools manages project variants and annotation sources for the user, it is easier to reanalyze variants as genomic builds change and as annotation sources are updated or become available. The burden of tracking VCF files, annotation files and numerous scripts is reduced. variant tools along with accompanying source code, documentation and tutorials are freely available at http://varianttools.sourceforge.net.
Sample and variant import: variant tools accommodates a variety of variant file formats. It supports import of VCF files or other tab-delimited formats such as intermediate output from ANNOVAR or BEDTools. It is capable of annotating and reporting on all types of variants, including indels, as long as annotation sources are available. The toolset also supports annotation and reporting of project variants using multiple genomic builds, by automatically downloading and integrating the UCSC liftOver tool (Hinrichs et al., 2006). As an example, if variants are imported to a project using build hg18, they can be annotated using annotation sources designed for build hg19, and exported based on either hg18 or hg19 coordinates.
Annotation: variant tools can incorporate databases that annotate individual variants or genomic regions, such as genes or pathways. A growing number of annotation sources such as dbNSFP (Liu et al., 2011) or KEGG pathways (Kanehisa and Goto, 2000) can be downloaded automatically whereas customized annotation databases could be created following a well-documented procedure. Any genomic data source can be imported into variant tools and used for annotations as long as variants can be linked to the new data through an existing variant attribute (such as genomic coordinates or a gene name).
Variant/sample selection (or filtering for quality control): variants or genotypes can be selected by read depth, by any of the available annotation fields (e.g. variant type, pathway or predicted damaging effects) or by sample frequency across subsets of samples, which is useful for comparing variants across populations (e.g. cases and controls). Complex criteria involving multiple fields from different annotation sources can be used to select or filter variants. Selected variants can be counted, exported or saved to separate underlying tables, where they can be annotated and filtered separately from other variants.
Report generation: variants can be exported in VCF or tab-delimited formats with an arbitrary number of fields. In addition to details provided by annotation sources, users can output sample statistics such as numbers of homozygous and heterozygous genotypes in samples for selected variants. Basic arithmetic operations and aggregate functions can also be used to output summary statistics of variants such as the average depth of coverage for a particular set of variants.
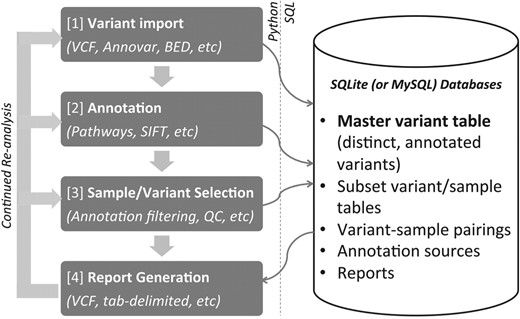
Example pipeline constructed from vtools utilities. variant tools provides a simple command-based interface that communicates with a back-end database. A large focus of variant tools is managing a master variant table that contains all the distinct variants of the project.
3 DISCUSSION
Despite an intuitive command-line interface, some high-level reports, such as calculating sample transition/transversion ratios or reporting the number of variants per gene, involve several vtools commands. To simplify the use of variant tools, we provide a reporting command vtools_report that generates example summary reports. These reports make the use of variant tools more practical, and the vtools_report source code provides examples of how to combine and further customize vtools commands.
Within variant tools, variants are linked to but stored separately from their annotations within a relational database, helping to conserve disk space by removing the need to store large and repetitive intermediate annotation files. Database indexes are automatically created to improve query performance during annotation and filtering, though these do add to the storage requirements of variant tools.
For an example, we created a vtools project with 44 whole-genome VCF files with 161 million predicted sample variants. This required 3.3GB of disk space to store the variants and indexes within an SQLite database compared to 2GB of disk space for the VCF files compressed or 9GB uncompressed. A benefit, however, of the vtools approach is that these variants were stored using both hg18 and hg19 genomic coordinates within SQLite. When using a MacPro workstation with two Quad-Core Intel Xeon Processors at 2.26 GHz and 8GB of RAM, the project creation required 3.5 hours. This time can be reduced to an hour if variants are processed in parallel by vtools on a cluster system before they are merged to a larger project. The time required for subsequent annotation and filtering of these variants ranged from 1 to 10 min. Additional details and other examples can be found in the tutorials section of the software website.
We have provided a preconfigured but customizable framework for the analysis of variants from next-generation sequence data. Although our efforts were motivated by a desire to produce initial, non-statistical analyses, we are currently expanding our software to include a suite of powerful tests for association studies. Our general framework will allow the implementation and comparison of a wide array of analytical methods.
Funding: National Institutes of Health (grants R01AR044422, U01 GM 92666, 5R03CA143982 and 1R01HG005859); Schissler Foundation (to F.A.S.L.); Lyda Hill Foundation (to B.P.).
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Alex Bateman


{kind=link}