Article Text

Abstract
AIM To study long term effects of interferon α2a (IFNα2a) on panuveitis in seven patients with Behçet’s disease in a prospective, open clinical trial.
METHODS Seven patients were treated with IFNα2a for a mean of 23.6 months (14–37 months). They received an initial dose of IFNα2a of 6×106IU/day, followed by 3×106 IU/day after 1 month and 3×106 IU every other day after 3 months. Two patients received low dose prednisolone (between 0.2 and 0.4 mg/kg/body weight) additionally at the beginning of the therapy. Complete cessation of IFNα2a was possible in three patients (observation period 22, 6, and 4 months).
RESULTS Marked improvement occurred in six patients who had ocular manifestations of Behçet’s disease for the first time or with minor damage during their course of chronic relapsing panuveitis. In one patient with advanced ocular Behçet’s disease, new relapses were prevented. Retinal infiltrates resolved within 2 weeks; vasculitis, macular oedema, infiltration of the anterior chamber and vitreous resolved within 4 weeks. Mean posterior uveitis score before treatment (nine affected eyes) was 6.6, 4 weeks after IFN it was reduced to 0.4. The mean observation period is 27.6 months, ranging from 14 to 42 months.
CONCLUSION Treatment of ocular symptoms of Behçet’s disease with IFNα2a alone or in combination with low dose steroids led to complete remission of ocular vasculitis in all patients treated in this open, uncontrolled trial. Treatment with IFNα2a may prevent permanent retinal or optic nerve damage due to vascular occlusion. No severe side effects occurred. Controlled randomised studies are warranted in order to prove the efficacy of IFNα2a in ocular Behçet’s disease and to compare it with other, established treatments such as azathioprine or cyclosporin A.
- Behçet’s disease
- uveitis
- interferon α2a
Statistics from Altmetric.com
Behçet’s disease is a multisystem inflammatory disorder. In addition to oral aphthosis, which is the hallmark of Behçet’s disease, frequent clinical manifestations comprise skin lesions, genital ulcers, arthritis, subcutaneous and deep vein thrombophlebitis. Ocular involvement occurs in 60–80%,1 2—in most cases a panuveitis with primary manifestation on average 8 years after disease onset and with a chronic relapsing course. Ocular manifestations are bilateral in most of the patients. Visual loss is mainly caused by retinal vasculitis involving superficial capillaries at the optic disc, macula, and retinal periphery and leading to occlusive vasculopathy. Perivenous and capillary leakage are the most common ocular findings, and sometimes only visible on fluorescein angiography.3 4
The central feature of the histopathology of Behçet’s disease is a systemic occlusive vasculitis (arteries and veins) with a tendency to venous thrombus formation.5 The underlying aetiology is not yet known. HLA-B51 association hints at a genetic component in the development of Behçet’s disease.6 7
The poor ocular prognosis of Behçet’s disease has improved over recent years with the increasing use of immunosuppressive agents.8 In particular, young male patients are at increased risk for ocular complications and require aggressive medical management. Azathioprine has been shown to maintain visual acuity (VA) and prevent the development of eye disease.9 Cyclosporin A is also an effective and rapidly acting drug for the treatment of eye disease in Behçet’s disease.10-12 Nephrotoxicity, particularly at doses higher than 5 mg/kg/day, relapses after cessation of therapy, and the high costs limit its use. Cytotoxic agents such as chlorambucil, and cyclophosphamide are also used but have been less well studied. Colchicine is effective for mucocutaneous and articular manifestations, but only partially effective for posterior uveitis.2 Brief courses of corticosteroids may shorten the duration of the attacks but they are not effective for long term treatment, probably because the dose necessary for maintainance of remission would be very high with unacceptable side effects.
Up to now, interferons have only been used in relatively small patient groups with Behçet’s disease. A few open studies with up to 20 patients excluding ocular disease, showed efficacy of IFNα and γ in different dosages.13-15 Recently, there have been case reports on four patients with severe refractory eye disease successfully treated with steroids, immunsuppressants, and IFNα in various combinations.16-18
Patients and methods
PATIENTS
We studied a total of seven patients. All were diagnosed as having Behçet’s disease according to the international study group criteria.19 First symptoms of the disease occurred approximately 3–14 years before ocular manifestation. All patients had clinical evidence of vision threatening retinal or optic nerve vasculitis. Each patient was admitted to hospital and, after initiation of IFNα therapy, examined daily for 10–14 days and then examined weekly for a period of 1 month. During remission patients were examined at intervals ranging from 1 to 3 months.
EVALUATION
All patients underwent ophthalmological examination including visual acuity, measurement of intraocular pressure, slit lamp examination of the anterior segment, and indirect ophthalmoscopy of the posterior segment. A general examination was performed at the department of internal medicine. Visual fields, fluorescein angiography, and fundus photography were performed at regular intervals, as well as laboratory tests including routine laboratory variables—erythrocyte sedimentation rate (ESR), C reactive protein (CRP), and autoantibody testing. Complete HLA typing was done at initiation of the therapy.
CRITERIA FOR EFFICACY
The uveitis scoring system20 which includes visual acuity, reduction of anterior and posterior activity scores, reduction of inflammatory activity in the fluorescein angiogram and reduction of inflammatory activity in the laboratory values (ESR, CRP) have been used as criteria for efficacy.
DOSAGE OF IFN α
IFNα2a 6×106 IU/day was administered initially. Depending on efficacy this dosage was tapered to 3×106 IU/day after 4–8 weeks and to 3×106IU every other day after 3–4 months. In one patient with Kaposi’s sarcoma, therapy was started with 18×106 IU/day, which is the clinical standard therapy for this disease. IFNα2awas always injected subcutaneously before bed time.
Concomitant low dose oral prednisolone in doses between 0.2 and 0.4 mg/kg body weight was given to two patients on steroid therapy at the time of exacerbation which could not be stopped promptly. Dosage was not changed in the first 2 months.
EXCLUSION CRITERIA AND CESSATION OF THERAPY
In case of inefficacy after 4 weeks or further deterioration, therapy was to be changed to CSA at the standard dosage of 3 mg/kg daily.11 12
Results
CASE REPORTS
Case 1
A 30 year old man of Turkish origin had suffered from Behçet’s disease since the age of 19. The first symptom was oral aphthosis; since June 1989 he had also had chronic relapsing panuveitis. Diagnosis of Behçet’s disease was made in February 1990. HLA-B51 was positive. Previous treatments were cyclosporin A (CSA, 5 mg/kg/day) plus low dose systemic steroids from July 1990 to November 1992, with several relapses of bilateral uveitis with persistent visual loss (chronic macular oedema, vascular occlusion, optic atrophy). In November 1992, azathioprine (AZA) 150 mg/day was added with consecutive complete remission of ocular inflammation. After 18 months of this triple immunosuppressive therapy the patient developed generalised Kaposi’s arcoma (stomach, small intestine, soft palate, skin, lungs). Thus, in April 1994 AZA and CSA were discontinued. Within days bilateral panuveitis with further visual loss occurred. VA was 0.02 in the right eye and 0.1 in the left. Ophthalmological examination revealed a bilateral hypopyon (R/L anterior chamber score 5) and segmental retinal vasculitis with a numerous scattered fresh infiltrates, macular oedema (retina score R 8/L 7) and marked vitreous infiltration (R/L score 3). Treatment with 3×106 IU IFNα2a was begun and increased to 18×106IU/day within 1 week (regular dosage for treatment of Kaposi’s sarcoma). Ocular inflammation resolved within 4 weeks. In October 1994 the dosage was tapered to 10×106 IU every other day. In February 1995 all lesions of Kaposi’s sarcoma had cleared. Since then, dosage has been reduced to 3×106 IU every other day. To date, no relapses of either disease have occurred (40 months). Visual acuity now is 0.2 (R) and 0.1 (L).
Case 2
A 31 year old woman of German origin had Behçet’s disease diagnosed in 1986. At that time a skin biopsy revealed a leucocytoclastic vasculitis. First clinical symptoms had occurred at the age of 17 (arthritis, oral aphthosis, and skin lesions). HLA-B51 was negative. Previously, from 1986 to 1990, interferon γ was successfully applied to treat mucocutaneous symptoms and arthritis.21 This case has been described in detail previously.18 In September 1993 she presented with an acute localised field defect in the right eye and ophthalmological examination revealed a large peripapillar retinal infiltrate in the left eye with otherwise normal fundus appearance in both eyes. VA was normal in both eyes. In the anterior chamber no cells or flare were detectable. There was a vitreous haze scoring 0–1 in both eyes but more pronounced in the left eye. Fluorescein angiography revealed a local blockage due to the nerve fibre oedema in the left eye without signs of vasculitis elsewhere. Systemic steroids were not effective (prednisolone 1 mg/kg), but when treated with higher doses of IFNα2a (6×106 IU/day), reperfusion occurred and the large retinal infiltrate resolved within 4 weeks. There was no loss of VA or persistent visual field defects. The patient was in stable remission on 3×106 IU IFNα2a twice weekly, which was tapered completely to zero by November 1995. No ocular relapses have occurred (22 months).
Case 3
A 30 year old woman of Greek origin had Behçet’s disease diagnosed in December 1994 at the time of her first ocular manifestation. At the age of 25 she already had the first symptoms of Behçet’s disease (oral aphthosis). Previous treatment consisted of CSA (4 mg/kg body weight) in December 1994 with good effect but was stopped by the patient after 3 months because of hirsutism. HLA-B51 was positive. At presentation in February 1995 with an acute onset of floaters and black patches in the visual field and pain of the right eye, together with a flare of oral and genital ulcers and arthritis of the right wrist, ophthalmoscopic examination revealed anterior chamber cell score and flare scores of 3 in the right eye. Vitreous haze scored 3. Funduscopically, there was a panuveitis with retinal vasculitis and retinal infiltrates in the mid periphery of the two lower retinal quadrants (posterior uveitis score 9) of the right eye. VA was reduced to 0.6 in the right eye. Fluorescein angiography disclosed vasculitis predominantly of the veins in the two lower quadrants and a mild macular oedema. The left eye was completely normal. IFNα2a therapy was started with 6×106IU/day. Retinal infiltrates resolved within 2 weeks and vasculitis within 4 weeks. Vitreous flare persisted between score 0–1. After 2 months, the dosage was reduced to 3×106 IU/day and after 6 months to 3×106 IU every other day. After reduction to 3×106 IU every other day a discrete relapse, manifesting as a localised area of vasculitis occurred, which disappeared after 2 weeks without increase of the IFNα2a dosage. In May 1997 IFNα2a was completely tapered to zero. VA is still 1.0 both eyes and visual fields are normal (4 months).
Case 4
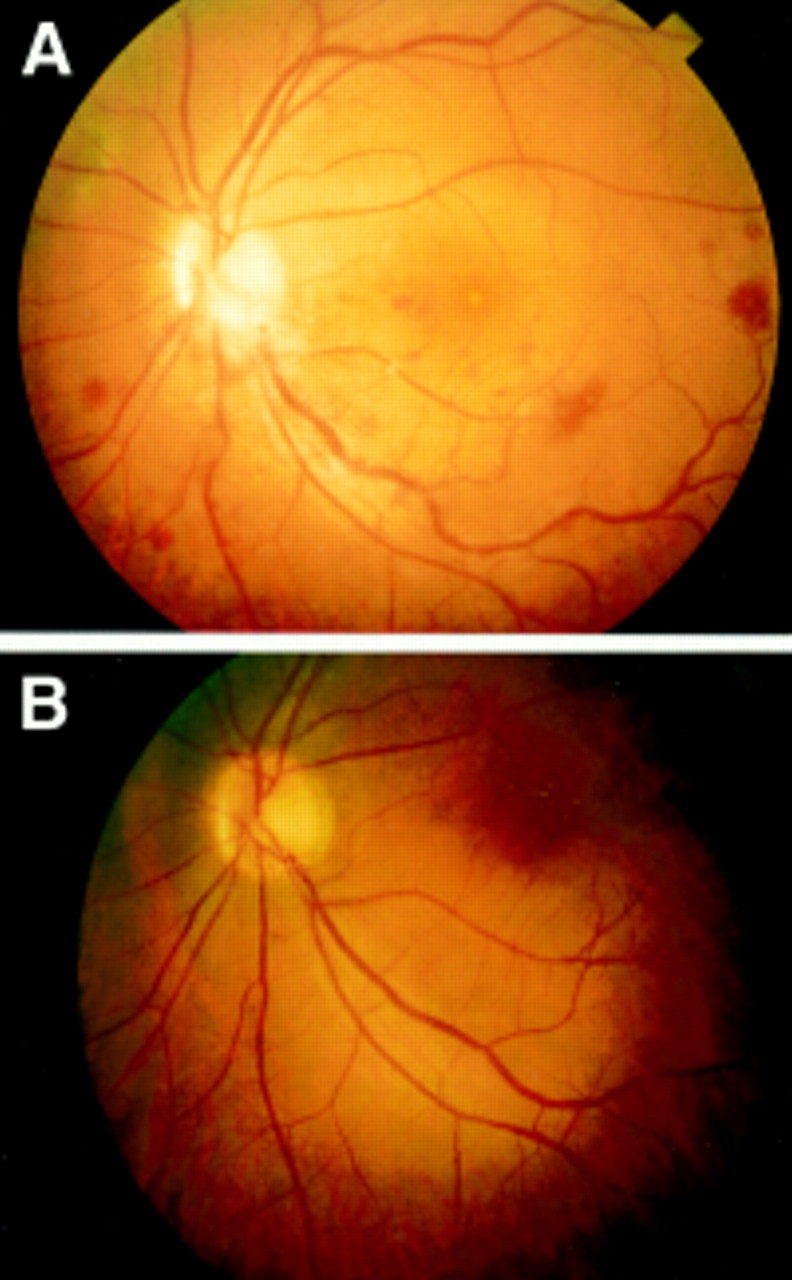
A 22 year old man of German origin had Behçet’s disease diagnosed in December1995. The first symptoms occurred at the age of 12 (oral aphthosis). Erythema nodosum lesions occurred for the first time in April 1995 and genital aphthosis in December 1995. HLA-B51 was positive. He was suffering from chronic relapsing posterior uveitis from February 1994, which was treated with prednisolone. Dose reduction below 10–20 mg/day caused recurrences of the uveitis. In December 1995 he presented with an acute reduction of VA, floaters, and visual field defects more in the right than in the left eye, together with oral aphthosis and dermal papulopustulosis. Ophthalmologically, VA was 0.2 the right eye and 0.8 in the left. Ocular examination revealed a mild anterior uveitis with anterior chamber cells scoring 1 in both eyes with no flare. Vitreous infiltration scored 2 in both eyes. The posterior uveitis score was 19 in the right and 2 in the left eye (Fig1A). Fluorescein angiography revealed active vasculitis.

Patient no 4 (A) Reconstruction of fundus photographs before treatment. Optic disc oedema, circular peripapillar flame-shaped haemorrhages, no central reflexes. Greyish appearance of the macula because of oedema is seen at the posterior pole. In all four quadrants in the periphery there are white infiltrates of the inner retina with blurred rims partially associated with intraretinal haemorrhages. There is diffuse scattered vasculitis most pronounced along the lower temporal branch vein with lower temporal branch stasis and marked intraretinal haemorrhages. (B) 14 days after treatment with 6×106 IU IFNα2a/day. Retinal infiltrates have completely resolved, optic disc and macular oedema regressed. Haemorrhages have partially resolved. Vascular sheathing persisted especially at the lower temporal branch vein, but there is clearly visible reperfusion of the lower branch vein. (C) 28 days after treatment with 6×106 IU IFNα2a/day. The optic disc is still minimally blurred. Macular oedema has resolved but central reflexes are irregular. Haemorrhages have markedly resolved. Vascular sheathing has disappeared except at a small lower temporal peripheral venule which still shows persistent occlusion and non-perfusion in the fluorescein angiogram.
Treatment was started in a general hospital with steroids beginning with prednisolone 100 mg which had been tapered to 40 mg when we first saw the patient. In our clinic therapy with IFNα2a(6×106 IU/day) was begun and the prednisolone dosage was reduced to 10 mg. This was maintained for the next 2 months. Retinal infiltrates resolved within 2 weeks (Fig 1B), and vasculitis disappeared within 4–6 weeks (Fig 1C). Fluorescein angiography disclosed almost normal venous filling after 2 months except for a small area of persistent occlusion of a lower temporal venule. Vitreous haze persisted between 0–1. VA improved to 1.0 both eyes. No visual field defects persisted. After 1 month, dosage was reduced to 3×106 IU/day; after 3 months to 3×106 IU every other day. The patient has been free of recurrence for 20 months.
Case 5
A 36 year old male patient of Italian origin had Behçet’s disease diagnosed in 1990 when he had orogenital aphthosis and arthritis of the right knee. The first symptoms had occurred at the age of 31 (oral aphthosis). HLA-B51 was positive. He had been effectively treated with IFN γ as far as the arthritic and mucocutaneous symptoms were concerned. This therapy was discontinued 2 years ago. In November 1995 he presented with acute reduction of VA and central field defects of the left eye together with dermal papulopustules, bilateral gonarthrititis, and orogenital apthosis. VA was 0.2 in the left eye and normal in the right eye. Ophthalmoscopic examination revealed an anterior chamber cell score of 3 and flare score of 1 in the left eye, but anterior chamber of the right eye was free of inflammation (score 0). Vitreous haze scored 2 in the left and 0 in the right eye. Funduscopy revealed papillitis with a disc oedema of 2 dioptres, nerve fibre layer bleeding at the rim of the disc, and sheathing of more than half of the vessels at the disc in the left eye (Fig 2A). There was retinal vasculitis with infiltrates and slight macular oedema with small hard exudates (retina score 7). Fluorescein angiography showed disc leakage and localised late staining of the vessels in the left eye (Fig 3A) but no changes in the right eye. Therapy with 6×106 IU IFNα2a/day was initiated. Because of marked, flu-like side effects and mental depression the dosage was tapered to 3×106 IU /day after 1 week. Retinal infiltrates resolved within 2 weeks, vasculitis disappeared within 6 weeks (Fig 2B). After 2 months, fluorescein angiography did not show any leakage of the disc and almost normal venous filling (Fig 3B). Vitreous flare persisted between 0–1. VA improved to 0.7 after 3 months of therapy. Currently, the patient has been free of any recurrence for 20 months.
Patient no 5 (A) Optic disc and posterior pole of the left eye before treatment with 3×106 IU IFNα2a/day. Optic disc shows hyperaemia with flame-shaped haemorrhages and marked oedema inferiorly. There is vasculitis of several veins of the two lower retinal quadrants. The lower temporal veins are tortuous and less oxygenated. (B) 28 days after treatment peripapillar haemorrhages and oedema have resolved. Collateral vessels have formed to the occluded lower branch vein, which now shows almost normal perfusion.

{kind=link}
{kind=link}
{kind=link}
Patient no 5 (A) Fluorescein angiography left eye mid transit (228 seconds) before treatment. There is irregular choroidal filling, delayed filling of the lower temporal branch vein, occlusive vasculitis leading to profound structural changes of the retinal vessels. (B) 8 weeks after initiation of treatment. There is relatively regular filling of the choroid and almost no delay of filling of the lower temporal vein partially due to the collateral vessels.
Case 6
A 38 year old man of Turkish origin had Behçet’s disease diagnosed in 1984, when he was suffering from recurrent panuveitis of both eyes. The first symptoms of Behçet’s disease occurred at the age of 16 (oral aphthosis). HLA-B51 was positive. He was treated with steroid pulse therapy several times, which was ineffective. At presentation in August 1996, he complained of blurred vision in the right eye for 2 months, together with oral aphthosis and dermal papulopustules. Ophthalmological examination revealed an anterior chamber cell score of 1 in both eyes with no flare. Fresh vitreous cells scored 1 in both eyes. In addition, there was marked vitreous clouding because of old cell detritus. Funduscopically, there was macular and disc oedema in the right eye and striking tortuosities of small arterioles in the retinal periphery which were leaking in the fluorescein angiography (retina score right eye 4, left eye 2). VA was 0.6 in the right eye and 1.0 the left eye.
He was treated with 3×106 IU IFNα2a/day for 1 month; later the dosage was tapered to 3×106 IU every other day because of nausea and diarrhoea. Macular and disc oedema resolved, VA increased to 1.0 again. After 3 months, IFN therapy was discontinued because of gastrointestinal symptoms. After 4 weeks a relapse of panuveitis in both eyes occurred together with marked oral aphthosis. VA was reduced to 0.5 in the right eye due to macular oedema. ESR and CRP increased. IFNα2a therapy (3×106 IU/day) for 1 week and then every other day was started again. Retinal lesions resolved, VA improved to 1.0 in both eyes again. The patient has been in complete remission for 17 months.
Case 7
A 27 year old woman of Turkish origin had Behçet’s disease diagnosed in May 1996; the first symptoms (cerebral vasculitis with hemiparesis and oral aphthosis) occurred in November 1989. HLA-B51 was negative. She had been treated with steroids several times, which were ineffective. At presentation in August 1996, she suffered from blurred vision in the left eye; ophthalmological examination of the left eye revealed an anterior chamber cell score of 1. Vitreous haze score was 2, and the posterior uveitis score was 10 (panuveitis with retinal vasculitis, retinal infiltrates in two quadrants, macular and disc oedema) in the left eye. VA was reduced to 0.6 in the left eye but was normal in the right eye. Fluorescein angiography revealed only mild optic disc vessel leakage in the left eye. IFNα2a therapy was initiated in August 1996 with 6×106 IU/day. Anterior chamber cells and flare resolved within 1 week, retinal infiltrates and retinal vasculitis improved within 1 month (posterior uveitis score 4). After reduction to 3×106 IU daily a further improvement occurred (November 1996 posterior uveitis score 2). In April 1997 IFN was tapered to zero because of the occurrence of antinuclear and anti dsDNA antibodies without clinically overt lupus erythematosus. The patient is in complete remission (6 months after discontinuation of IFN) with a visual acuity of 1.0 (R/L).
The patients’ data are summarised in Table 1, the results of the ophthalmological examinations in Table 2.
Summary of clinical and patient data
Visual acuity and uveitis scores before and 4 weeks after (in parentheses) initiation of IFNα2atherapy
In summary, in all seven patients, ocular manifestations of Behçet’s disease entered complete remission after treatment with IFNα2a. In five patients (nos 2, 3, 5, 6, 7) IFNα2a monotherapy was effective. In another two combination with 2–10 mg of prednisolone daily was required (patients 1 and 4). Under IFNα2a therapy, retinal infiltrates resolved after approximately 2–3 weeks in all patients, and active vascular sheathing disappeared after approximately 4–6 weeks. Most of the occluded vessels were reperfused, and only localised small areas of non-perfusion persisted. Vitreous opacity persisted longer but signs of active disease disappeared after 4 weeks. Anterior segment inflammation, additionally treated with local steroids, completely resolved within 4 weeks.
In the patients who had a short history of ocular involvement with good visual acuity before outbreak or relapse of the uveitis only minor damage persisted (patients 2–7).
Five patients have had no relapses up to now (patient 1, duration of remission 40 months; patient 2, 22 months; patient 4, 20 months; patient 5, 20 months; patient 7, 6 months). Patients 3 and 6 relapsed 4 weeks after cessation of relatively low dose IFN therapy or after dose reduction below 3×106 IU IFNα2a every other day. Both patients were in complete remission again after augmentation of dosage. In three patients (nos 2, 3, 7) IFNα2a could be tapered to zero without relapse of uveitis up to now (observation period since cessation of therapy 22, 4, and 6 months, respectively). Mean posterior uveitis score before treatment (nine affected eyes) was 6.6; 4 weeks after initiation of IFNα2a therapy it was reduced to 0.4. The mean observation period is 27.6 months, ranging from 14 to 42 months.
Discussion
All seven patients treated with IFNα2a entered complete remission of ocular vasculitis/panuveitis. In three of the patients, complete cessation of IFN therapy was possible without relapse of uveitis. An effective maintenance dose of IFNα2a seems to be 3×106 IU daily with the possibility of complete cessation of therapy in at least some of the patients.
It may be that some of the remissions in our patients were the result of the natural undulating course of Behçet’s disease, but the close temporal relation of remissions to initiation of IFN therapy and relapses in some patients to dose reductions strongly suggests a therapeutic effect of IFNα2a.
Long term treatment (five patients now for more than 2 years) did not cause serious side effects and in this respect IFN may be superior to immunsuppressants. In all patients flu-like symptoms following the first injections were treated with paracetamol. With 6×106 IU/day and above, slight thrombocytopenia and leucocytopenia occurred. However, patient 1 tolerated the high dose of 18×106 IU/day very well. All patients experienced a mild alopecia and reddening at the site of injection. One patient had moderate gastrointestinal side effects (diarrhoea) and one patient had temporary mental depression; one patient developed antinuclear antibodies and dsDNA antibodies and one antithyroid antibodies without clinically overt autoimmune disease.The induction of autoantibody production by IFNα and even autoimmune disease, especially autoimmune thyroid disease, has been observed in patients with chronic hepatitis C or malignant haematological diseases.22 Another, less frequent side effect described in literature is an interferon induced retinopathy with retinal infiltrates similar to those occurring in Behçet’s disease itself, which also mainly has been observed in patients with chronic hepatitis23 24 and an anterior ischaemic optic neuropathy.25 The development of cutaneous leucocytoclastic vasculitis and even Behçet’s disease itself during IFNα treatment has been described.26-28
We did not observe retinopathy, vasculitis, or worsening of Behçet’s disease in our patient group during the observation period of 5 years. This, of course, could be due to the small number of patients treated or the shortness of the observation period. In our opinion, the development of retinopathy in hepatitis C could possibly be explained by autoimmune phenomena due to the chronic viral infection. A retinopathy also exists in chronic myelogenous leukaemia and hairy cell leukaemia as a disease related phenomenon. Thus, studies comparing patients with hepatitis C without interferon treatment are necessary to evaluate whether retinopathy really is more frequent in patients on IFNα treatment. We also believe that the development of Behçet’s disease during IFNα treatment for chronic myelogenous leukaemia (CML) either may be hazardous, because the three cases described by now were all observed in areas where Behçet’s disease is endemic (Japan and Turkey) or the result of disease specific reactions in CML—for example, a specific change in adhesion status of the CML clones which, in genetically susceptible individuals, consecutively leads to symptoms of Behçet’s disease.
One rationale for using interferons in Behçet’s disease is their efficacy in other immune complex associated vasculitides, in which an infectious trigger is known—for example, cryoglobulinaemia (hepatitis C) and polyarteritis nodosa (hepatitis B). The possibility that viral infections may have an aetiological role in Behçet’s disease has been postulated previously by several investigators. The microbiological agents proposed include herpes simplex virus29-31 and parvovirus B19.32 Bacteria, mainly strains of streptococcus, have also been suggested as candidate infectious agents.33 Recently, hepatitis C virus was implicated in the aetiology of Behçet’s disease, but preliminary results of one group were not proved by another.34 35 Of our patients, none was serologically positive for hepatitis C (results not shown).
Additionally, there is evidence for a polarisation towards the Th1 functional profile in Behçet’s disease.36 37 A substitution with IFNα further diverts the T cell response in the direction of Th1.38 This and some other immunomodulatory actions of IFNα as enhancement of HLA class I antigen expression on lymphoid cells and of T and NK cell cytotoxicity may be helpful in improving elimination of foreign antigens.
We therefore conclude that IFNα2a alone or in combination with low dose steroid therapy may be effective in treating ocular Behçet’s disease. Of note, randomised controlled studies are necessary to further prove this. Optimal dosage also has to be evaluated. Our current recommendation is 6×106 IU IFNα2a/day for the first month with consecutive tapering to 3×106 IU /day, later every other day to three times weekly. The most frequent side effects are flu-like symptoms, mild alopecia, and development of autoantibodies without clinically overt autoimmune disease.
Acknowledgments
We thank Dr Graham Pawelec for his critical review of our manuscript.