Article Text

Abstract
AIMS/BACKGROUND The purpose of this study was apply the polymerase chain reaction (PCR) to develop a sensitive, specific, and rapid test to diagnose Fusarium keratitis.Fusarium is the most common cause of fungal corneal infection in some parts of the world. It is often difficult to establish that a keratitis is due to fungal infection.
METHODS Fusarium solanikeratitis was induced in three eyes of three rabbits by injection of a suspension of the fungus into the anterior corneal stroma. In one rabbit the contralateral eye served as a control. From four to 28 days after inoculation, the corneas were scraped for culture, then scraped and swabbed for PCR analysis. The PCR was performed with primers directed against a portion of the Fusarium cutinase gene, and the presence or absence of this amplified target sequence was determined by agarose gel.
RESULTS The amplified DNA sequence was detected in 25 of 28 samples from the corneas infected with Fusarium, for a sensitivity of 89%. Only three of the 14 samples from these eyes with Fusarium keratitis were positive by culture, for a sensitivity of 21%. Seven of eight control samples were negative by the PCR based test, for a specificity of 88%.
CONCLUSION This PCR based test holds promise of being an effective method of diagnosing Fusarium keratitis as well as Fusarium infections at other sites.
- keratitis
- Fusarium
- ulcer
- cornea
- polymerase chain reaction
Statistics from Altmetric.com
In some parts of the world, fungal infection accounts for over one third of corneal ulcers.1 Fusarium, a genus of filamentary fungus, is the most common cause of fungal keratitis in parts of the southern United States and Africa.2-8 Corneal infections with filamentary fungi occur predominantly in healthy, young men with a history of outdoor, ocular trauma.1-4 8 9
The diagnosis of fungal keratitis continues to be problematic. Fungal ulcers can have feathery, hyphate edges, raised borders, a white or greyish colour, satellite lesions, and an associated endothelial plaque.3 8 9 However, many fungal ulcers demonstrate no striking morphological pattern.1 10 Since many of these characteristics are not specific to fungal ulcers, the literature recommends that antifungal therapy be withheld until diagnosis is confirmed by laboratory studies.7 9 Gram and Giemsa stains of corneal scrapings have sensitivities of about 50% in establishing the diagnosis.1 3 4 8 11 Fungal cultures from corneal scrapings often take 3 to 4 days, and can take weeks, to become positive.1 8 11 Culture has been used as the “gold standard” for the diagnosis of fungal keratitis, so true sensitivity of culture is unknown.4 8 The laboratory diagnosis of fungal keratitis may be problematic because of the very small sample which can be obtained by scraping a corneal ulcer.
The polymerase chain reaction (PCR) technique permits the in vitro copying and recopying of a selected DNA sequence in a series of denaturation, reannealing, and extension steps in the presence of synthetic oligonucleotide primers and heat stable DNA polymerase.12 13 This allows amplification of a target sequence to concentrations which are easily detectable by conventional laboratory methods. When applied to the detection of infection, the PCR is extremely sensitive and can establish the presence of even a few organisms in a sample. The amplification occurs if and only if the target DNA sequence is in the reaction mixture. Therefore, with careful selection of the target sequence, the PCR can be a highly specific test. A PCR based test can be accomplished in a few hours, and so is much more rapid than culture. PCR amplification has been successfully applied to the diagnosis of viruses, bacteria, and protozoa from the ocular surface, corneal buttons, aqueous samples, and vitreous samples.14-23
We have developed a PCR based test which detects a portion of theFusarium genome and have applied it to confirm the diagnosis of a Fusarium panophthalmitis in postmortem tissues.24 In this study, we investigated the potential for use of this PCR based test in the diagnosis of activeFusarium keratitis. We applied the test to samples collected from experimentally induced corneal ulcers in rabbits.
Materials and methods
ANIMAL MODEL OF FUSARIUM KERATITIS
Two strains of Fusarium solani, isolated from patients with fungal keratitis were obtained as lyophilised pellets (strains S-432, S-446; Fusarium Research Center, Penn State, University Park, PA, USA). The fungi were passaged on Sabouraud’s dextrose agar plates at room temperature, and an inoculum was prepared by suspending harvested spores in sterile yeast peptone broth.
Adult, male New Zealand albino rabbits were anaesthetised with an intramuscular injection of 5 mg/kg xylazine and 40 mg/kg ketamine. Using a portable slit lamp, a syringe with a 30 gauge needle was inserted tangentially into the central corneal stroma to a depth of about one third of the corneal thickness.25 About 50 μl of the inoculum, the equivalent of 50 000 Fusariumspores, were injected.
A Fusarium solani keratitis was induced in one eye of each of three rabbits. Two of these eyes also received 1.0 mg of subconjunctival triamcinolone acetonide subconjunctivally at the time of inoculation. The contralateral eye of one of these rabbits served as a control. It was first inoculated with sterile peptone broth, and 14 days later a Candida albicans corneal ulcer was induced as described for Fusarium. All animal research was approved by an institutional review and conformed with the guidelines of the Yale University Animal Use and Care Committee.
SAMPLE COLLECTION AND PREPARATION
Sample collection was performed four or five times during the period from 4 to 28 days after inoculation. The rabbits were anaesthetised as described above. A Kimura spatula was thoroughly flamed and then cooled with sterile water before each scraping. Using the portable slit lamp and the spatula, the base of the corneal ulcer (or the injection site in the case of the control eye to the end of day 12) was scraped using the Kimura spatula. The material obtained was minimal and in most samples not visible on the spatula tip. The cornea was initially scraped for culture on Sabouraud’s dextrose agar plates with gentamicin. The cornea was then scraped for the PCR, and the spatula was stirred for a few seconds in 150 μl of deionised, sterile water in a 1.5 ml sterile Eppendorf tube. Finally, the cornea was swabbed for PCR using a Rayon swab. The swab was placed in 150 μl of deionised, sterile water in a 1.5 ml sterile Eppendorf tube. The Sabouraud’s plates were incubated at room temperature for 4 weeks.
All samples obtained from the rabbit corneas for PCR were stored at −20°C for up to 4 weeks until processing. They were initially thawed to room temperature. In order to release DNA from any spores, the entire sample was heated in the thermal cycler (Ericomp, San Diego, CA, USA) at 100°C for 15 minutes, and then the specimens were chilled on ice. The swabs were discarded from those samples obtained with swabs. Three aliquots, 1–35 μl, were taken from each sample and each aliquot placed directly into a separate PCR mixture.
POLYMERASE CHAIN REACTION PROTOCOL
The oligonucleotide primers used for the PCR12 were sequences complementary to a 189 base pair portion of theFusarium solani cutinase gene.26 27 The primers had the following sequences:
5′ -ATC GAG GAC CTC GAC TCG- 3′
5′ -GCA GCA ACG ATC AAG CTA- 3′
Each 50 μl PCR mixture contained 10 mmol/l TRIS-HCl (pH 8.4), 50 mmol/l KCl, 1.5 mmol/l MgCl2, 200 mmol/l of each of all four deoxyribonuleoside triphosphates, 2.5 units of Taq polymerase (Promega), 75 pmol each of the oligonucleotide primers, and an aliquot from the corneal samples.
Using the “hot start” method to improve specificity, samples were kept at 80°C for 2–3 minutes and then the deoxyribonuleoside triphosphates were added to each sample.28 Samples were initially denatured for 7 minutes at 94°C, and then subjected to 37 cycles of amplification using the following variables: denaturation at 94°C for 60 seconds, annealing at 55°C for 70 seconds, and extension at 72°C for 100 seconds. After the last cycle, samples were incubated for 10 minutes at 72°C. Standard methods were followed in order to avoid false positive results caused by PCR product carryover.29
A positive and negative control were included for every set of PCRs. For the positive control, the sample consisted of the purifiedFusarium solani DNA extracted from fresh mycelium (see below). For the negative control, the sample consisted of deionised, sterile water.
DETECTION OF THE PCR PRODUCT
A volume of 8 μl of each amplification reaction was analysed by electrophoresis on a 1.8% agarose gel and the DNA fragments were visualised by staining with ethidium bromide. If the positive and negative controls did not yield the expected results, the results of the run were considered invalid, and the run repeated.
The presence or absence of the target DNA fragment after PCR amplification was also confirmed using Southern blot hybridisation with a 5′ biotin labelled internal probe.14 30 The probe had the following sequence 5′ -AGATCGCCGGAACTGTTCTGTTCGGC TACA- 3′. The product was detected using a streptavidin-alkaline phosphatase conjugate dephosphorylation reaction system.
Restriction mapping of the amplified target sequence DNA was performed by digesting the purified 189 base pair PCR product with the restriction endonucleases Sty1, BsaJ1, or EcoO109I. The digestion products analysed on agarose gel.
DNA PREPARATION FOR CONTROL SAMPLES
DNA was extracted from fresh mycelium grown on Sabouraud’s dextrose agar plates by lysis for 1 hour at 65°C in a 50 mM TRIS-HCl (pH 7.2) buffer containing 50 mM EDTA, 3% SDS, and 1% 2-mercaptoethanol. The DNA was purified by phenol-chloroform extraction repeated three times, followed by ethanol precipitation. The pellet was suspended in deionised, sterile water and aliquots were used as a positive control for each PCR run.
Purified DNA samples from other species causing infectious keratitis were isolated in order to determine the specificity of the PCR based test. Cultures of other fungi, bacteria, and viruses which commonly cause infectious keratitis were well characterised clinical isolates provided by a clinical department of microbiology. ForFusarium oxysporum (strain O-783, Fusarium Research Center, isolated from a patient with mycotic keratitis),Aspergillus fumigatus, Penicillium, and Candida albicans, DNA was extracted and purified as described above. DNA was extracted from Staphylococcus aureus, Pseudomonas aeruginosa, Streptococcus pneumonia, and herpes simplex, as previously described.14 19 In addition, a scraping from human buccal mucosa was processed in the same manner as the rabbit cornea scrapings.
Results
ANIMAL MODEL OF FUSARIUM KERATITIS
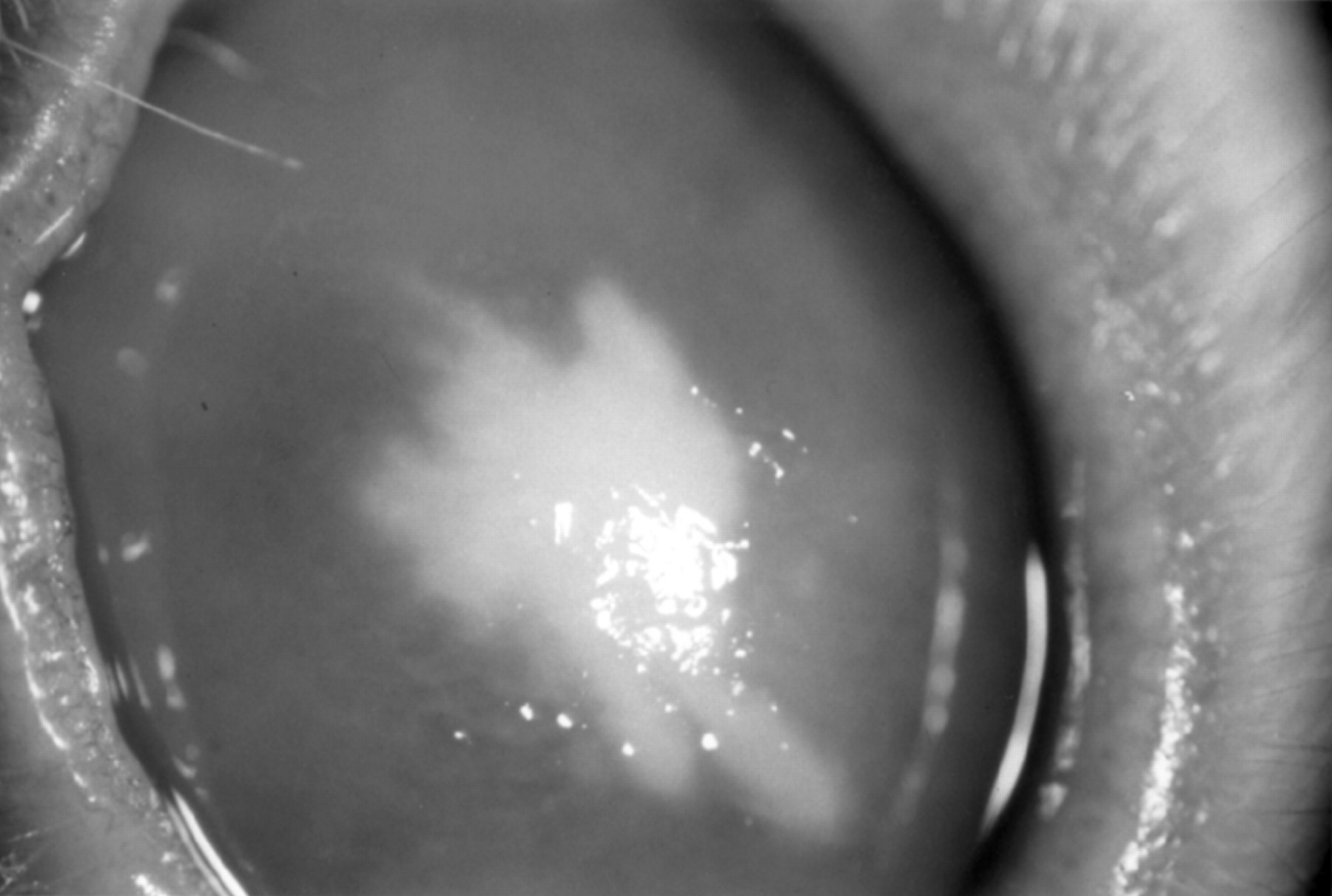
Using the procedures described above, severe Fusarium solani infections were apparently induced. Two to 4 days after inoculation with Fusarium, a 2–4 mm central infiltrate appeared at the inoculation site, and the peripheral cornea became oedematous (Fig 1). By day 8 peripheral vascularisation began. By day 12 the stromal infiltrate had progressed to the surface to produce an epithelial defect with some stromal thinning, and the vessels had grown into the infiltrate (Fig 2). By days 24–28 the infiltrate was subsiding, and a 3–4 mm vascularised scar began to form. The injection of subconjunctival triamcinolone at the time of inoculation delayed the inflammatory signs for a day or two, but the subsequent inflammation was greater than in the eye in which no corticosteroid was used.

A rabbit cornea 5 days after inoculation with Fusarium solani, showing a focal infiltrate and diffuse corneal oedema.

A rabbit cornea 12 days after inoculation with Fusarium solani, showing vascularisation of the infiltrate.
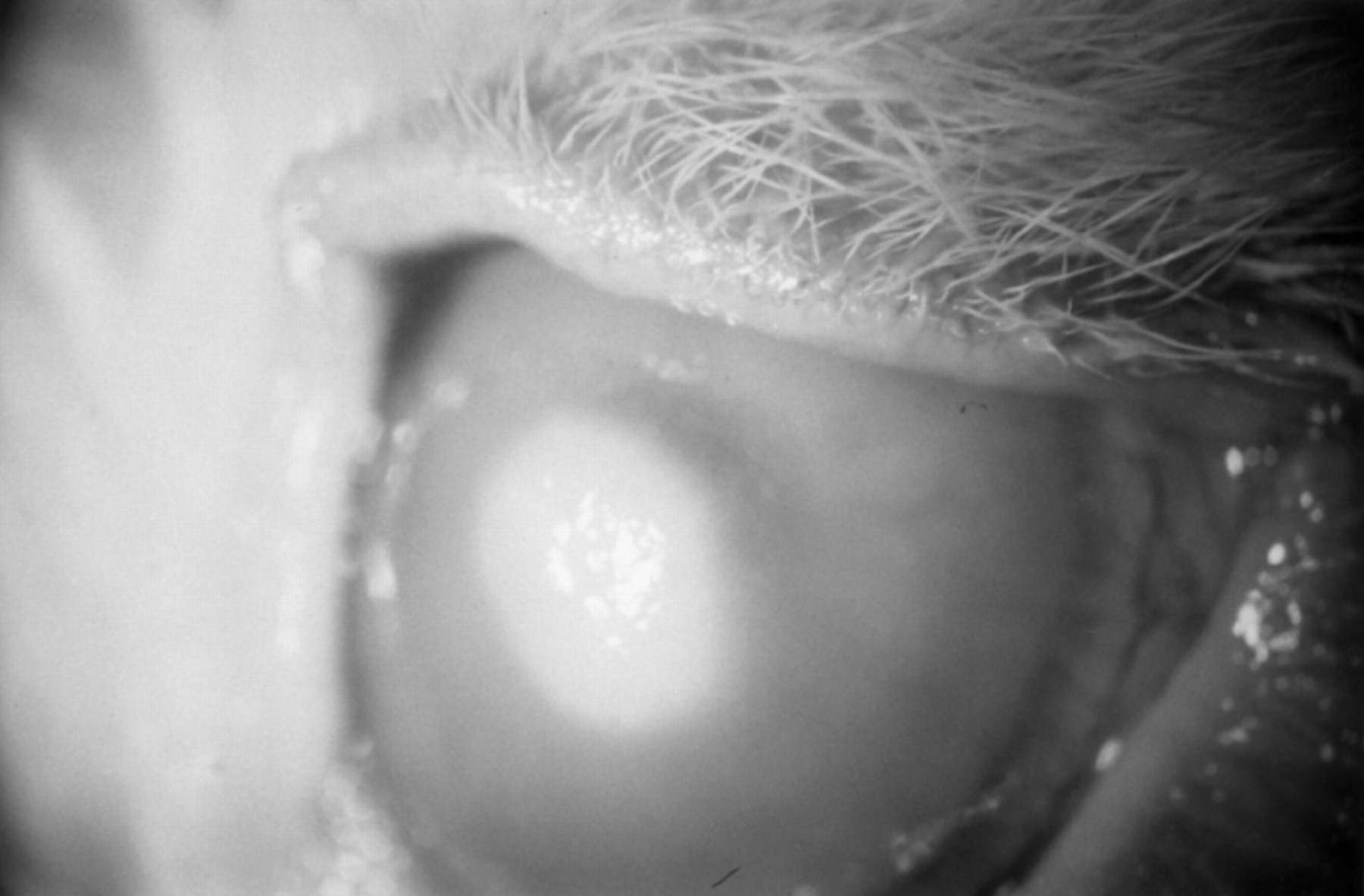
Injection of the control eye with sterile peptone broth produced no inflammatory signs. When the same eye was subsequently inoculated withCandida albicans, inflammatory signs, similar in their course to that described for Fusarium, ensued (Fig 3).
A rabbit cornea 4 days after inoculation with Candida albicans, showing a focal infiltrate and diffuse corneal oedema.
PCR AND CULTURE RESULTS
Twenty eight PCR samples (15 scrapings and 13 swabs) were obtained from corneas inoculated with Fusarium solaniand eight samples (four scrapings and four swabs) were obtained from the control eye. A sample was considered positive by the PCR method if and only if one or more of the three aliquots yielded a PCR product visible as a band of expected size on agarose gel electrophoresis (Fig4).

Agarose gel showing PCR results of samples collected by scraping and swabbing Fusarium infected and control corneas. Many of the positive results from the Fusarium infected eyes consist of only faint bands of the target fragment of the cutinase gene, which are lost on reproduction of the photograph. However, the faint bands were confirmed by Southern blot analysis. As expected, the PCR products from the control eye contained no detectable target DNA. A separate negative control was from the PCR performed at the same time with sterile water substituted for the sample. The positive control was PCR product from purified DNA extracted from fresh Fusarium mycelium.
The presence of the target DNA was detected in 25 of the 28 samples from corneas inoculated with Fusarium solani(Table 1). Assuming that all samples collected fromFusarium inoculated corneas were infected withFusarium when the samples were collected, the sensitivity of the technique was 89%. Scrapings and swabs gave similar results. Target DNA was also amplified in one of the eight control samples, for a specificity of 88%. All negative results and all but two positive results by agarose gel were confirmed by Southern blot analysis.
Summary of culture and polymerase chain reaction (PCR) results
Only three of the 14 cultures of scrapings from Fusariuminfected corneas plated on Sabouraud’s dextrose agar grewFusarium, for a sensitivity of 21%. All positive cultures were from within the first 13 days after inoculation of the cornea. For each positive culture, fungal colonies grew within 3 days of sample collection. For all samples that were positive by culture, the corresponding PCR samples were both positive. There were no false positive cultures, for a specificity of 100%.
LIMITS OF DETECTION AND SPECIFICITY
The identity of the 189 base pair PCR amplification product was confirmed by restriction endonuclease digestion. The restriction enzymes produced fragments of the expected sizes (Fig 5).

Agarose gel of restriction endonuclease digests of the PCR product. Fragments of the expected sizes were produced (Sty1: 122 base pair (bp) and 67 bp; BsaJ1: 87 bp, 67 bp, and 35 bp; EcoO109I: 174 bp and 15 bp), confirming that the product was the target fragment of the Fusarium cutinase gene.
In order to establish the limits of detection of this PCR technique, amplifications of serial dilutions of Fusarium solani spores suspended in deionised, sterile water were performed. Using the same sample preparation and PCR protocols as those described for the rabbit corneal specimens, the PCR was able to amplify the fragment of the cutinase gene from as few as 10Fusarium spores to levels detectable by agarose gel (Fig6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Agarose gel of PCR products from a serial dilution of Fusarium solani spores, demonstrating that this PCR based test can detect from 10 to 10 000 organisms in a sample. Positive and negative controls are as described in Figure 4.
Homologies between the cutinase gene primers and other known DNA sequences were excluded by a computer analysis of all sequences available in GenBank (June 1993; Genetics Computer Group, Madison, WI, USA). Specificity of the PCR assay was further tested by conducting PCR amplifications with purified extracted DNA from non-Fusarium samples, using the cutinase gene primers and the same PCR protocol. DNA isolated from Aspergillus fumigatus, Penicillium, Candida albicans, Staphylococcus aureus, Pseudomonas aeruginosa, Streptococcus pneumonia, and herpes simplex failed to result in the amplification of the 189 base pair fragment by both agarose gel analysis and Southern blot hybridisation. A scraping from human buccal mucosa, which contained human cells (and probably a variety of bacteria), also yielded negative results. The PCR test did amplify the target sequence when performed with purifiedFusarium oxysporum DNA extracted from mycelium.
Discussion
In this preliminary study, our PCR based test was evaluated for its efficacy in the detection of Fusarium keratitis from corneal scrapings and swabs in an animal model of fungal keratitis. If it is assumed that the corneas inoculated with Fusarium solani were infected with the organism at all times that samples were collected (our “gold standard”), the PCR based test was significantly more sensitive than culture in the detection ofFusarium infection. The PCR based test had a sensitivity of 89%, compared with 21% for culture.
One explanation for this difference is that a culture is positive only if the sample contains viable organisms, while a PCR based test will detect both viable and non-viable organisms. A PCR test can be positive even if only a single copy of the target DNA is present.31
In studies of human keratitis, about one third of all cultured corneal ulcers are culture negative,1 32 so it is unknown how many fungal ulcers go undiagnosed by currently available culture techniques. Blood agar, Sabouraud’s media, and thioglycolate are the only media commonly used in the examination of ulcers that are likely to yield positive fungal cultures. The combination of these three media had a sensitivity of less than 50% among culture positive fungal corneal ulcers in a study that used additional media.11Although many fungal cultures from corneal infections can become positive within 72 hours of sample collection, about one fourth become positive only after 2 weeks.1 8 11 Among the cases of human fungal keratitis, Gram and Giemsa stains have sensitivities of only 25% to 66% in different series.2 3 4 8 11 Given the sensitivity of the PCR test and the fact that it could be completed within 4 hours of sample collection, the test has the potential to become a very useful test in the diagnosis of Fusarium.
Although the PCR based test was four times more sensitive than culture, three samples from Fusarium infected corneas were negative. All false negative samples were obtained from the same corticosteroid treated Fusarium eye during the period of 4 to 9 days after inoculation. Since corticosteroid administration suppressed the clinical response to infection by filamentary fungi,33 it is possible that the infection was not sufficiently superficial to obtain an adequate sample. Another possibility is that the inflammatory response to the infection damaged the Fusarium DNA or otherwise inhibited the PCR.
The specificity in our series was only 88%, due to a single false positive. Contamination from the contralateral Fusariuminfected eye is the most likely explanation for the false positive. Although standard methods were followed during all procedures in order to avoid PCR product carryover,29 contamination during the preparation of the PCR samples is another possibility.
We chose a fragment of the cutinase gene as our target DNA because we felt it could be specific to Fusarium but conserved within the genus. Cutinases are excreted by phytopathogenic fungi and catalyse the hydrolysis of the structural polyester of the plant cuticle and so are important to the survival of the fungus.26 27 The test was able to detect Fusarium oxysporum as well as Fusarium solani. We also demonstrated that applying the Fusarium PCR based test to other corneal infectious agents, including other fungi, bacteria, and herpes simplex, yields negative results.
In this study, all specimens obtained from Fusariuminfected eyes during the fourth week after inoculation were positive for the target DNA, although the corneal ulcers appeared to be nearly healed and cultures were negative. This may be an illustration of the fact that PCR based tests for infection will be positive whenever the sample contains a sufficient number of copies of the target DNA, whether the organisms are dead, constitute normal surface flora, or are causing an active infection.
Our technique involved only boiling of the PCR samples and adding aliquots directly to the PCR mixture without any purification steps. This is a convenient and rapid method of DNA preparation, reduces the risk of contamination, and eliminates the DNA loss which would occur from attempting DNA purification. Releasing DNA from filamentary fungi is problematic since these organisms have resilient cell walls. Boiling for 15 minutes apparently releases the fungus DNA without damaging the cutinase gene sufficiently to inhibit the PCR. Further refinement of the sample preparation for the PCR may improve the sensitivity of the test.
This PCR based test holds promise to be an effective method of diagnosing Fusarium keratitis in the clinical setting. Compared with standard laboratory techniques, it offers increased sensitivity as well as a significant reduction in the time required to establish the diagnosis. However, further studies are needed to refine the technique, improve its sensitivity and specificity, and to establish the value of the technique in managing patients with corneal ulcers. Systemic fungal infection is becoming an increasingly common infection among iatrogenically immunosuppressed patients, where the diagnosis is also problematic.34-37 Therefore, the technique may eventually have wide application in the diagnosis of fungal disease.
Acknowledgments
Presented at the Association for Research in Vision and Ophthalmology annual meeting, Sarasota, FL, USA, 3 May 1994.
This study was supported by a student fellowship award, in memory of Norma Newman Cohen, from the Fight for Sight Research Division of the National Society to Prevent Blindness (GA) and a grant from Research to Prevent Blindness (PG).
The authors thank Sandy Waycott at the Department of Microbiology of Yale University for providing the clinical isolates. They thank John Danias, MD, PhD, and Iqbal Ahmad, PhD, for their technical advice. They also thank Marvin L Sears, MD for providing the laboratory facilities.
References
Footnotes
-
↵Current address: Bascom Palmer Eye Institute, University of Miami School of Medicine, Miami, FL 33101, USA.