Article Text

Abstract
AIM To report a Japanese family diagnosed clinically as having lattice corneal dystrophy type I (LCDI) in which a Leu518Pro mutation in the βig-h3 gene and not the R124C mutation reported previously was found.
METHODS Molecular genetic analysis was performed on DNA extracted from peripheral leucocytes from four members (three affected and one unaffected) of a family. Exon 4 of the βig-h3 gene was amplified by PCR and directly sequenced. Histopathological study was performed on the corneal tissue from the proband obtained during deep lamellar keratoplasty.
RESULTS All the affected members were clinically diagnosed as having LCDI, and the pedigree indicated an autosomal dominant inheritance. A heterozygous single base pair transition (CTG to CCG, leucine to proline) was detected in codon 518 of the βig-h3 gene in the three affected members, and not in the unaffected member. No mutation was found in codon 124. Amyloid deposits were observed between the collagen bundles of the corneal stroma and were seen to extend deep into the stroma.
CONCLUSION The Leu518Pro mutated βig-h3 forms amyloidogeneic intermediates which precipitate in the cornea and gives rise to a clinical appearance of LCDI.
- corneal amyloidosis
- lattice corneal dystrophy type I
- βig-h3 gene
- codon 518
Statistics from Altmetric.com
Lattice corneal dystrophy type I (LCDI) is an autosomal dominant bilaterally symmetric corneal disorder which is characterised by numerous translucent lattice lines associated with white dots and faint haze in the superficial and middle layers of the central stroma.1 2 The symptoms appear during the first or second decades of life.1 2 This dystrophy, along with another three autosomal dominant corneal disorders, granular corneal dystrophy Groenouw type I, Avellino corneal dystrophy, and Reis-Bücklers corneal dystrophy, has been mapped to chromosome 5q31.3-5 Recent studies have shown that these four dystrophies result from the mutation of TGF-β induced gene (βig-h3),6 and a R124C mutation was detected in the LCDI in both white6 and Japanese7 families. Recently, Endo et al found a new mutation of βig-h3 in a Japanese family clinically diagnosed as LCDI in which a mutation could not be found in R124 but instead a Leu518Pro mutation was detected.8 We present another Japanese family with the Leu518Pro mutation. Histopathological examination of a corneal specimen from one of the patients showed amyloid deposits between the collagen bundles of the corneal stroma.
Patients
The pedigree being studied is shown in Figure 1.

Pedigree of the family. The arrow indicates the proband (III-4). III-7 and 8 are fraternal twin sisters.
The proband (III-4) is 42 year old man who was referred to the department of ophthalmology of the Nagoya University Hospital from an internist. The patient complained of visual disturbance during his stay in the same hospital as an inpatient for renal failure of unknown cause. He first suffered ocular pain at 15 years of age, and noticed a visual acuity loss at 20 years of age. He was checked by an ophthalmologist and his visual acuity gradually decreased. On the first visit, his visual acuity was 20/200 non-correctable (NC) in the right eye, and 20/1000 NC in the left. The intraocular pressure (IOP) was 11 mm Hg right eye and 10 mm Hg left eye. A dense superficial corneal opacity associated with fine lattice lines in the superficial and midstromal layers was observed in the both eyes by slit lamp biomicroscopic examination (Fig 2a). The epithelium appeared smooth without any fluorescein stained regions. The lens appeared transparent but the ocular fundus was not visible through the hazy cornea.
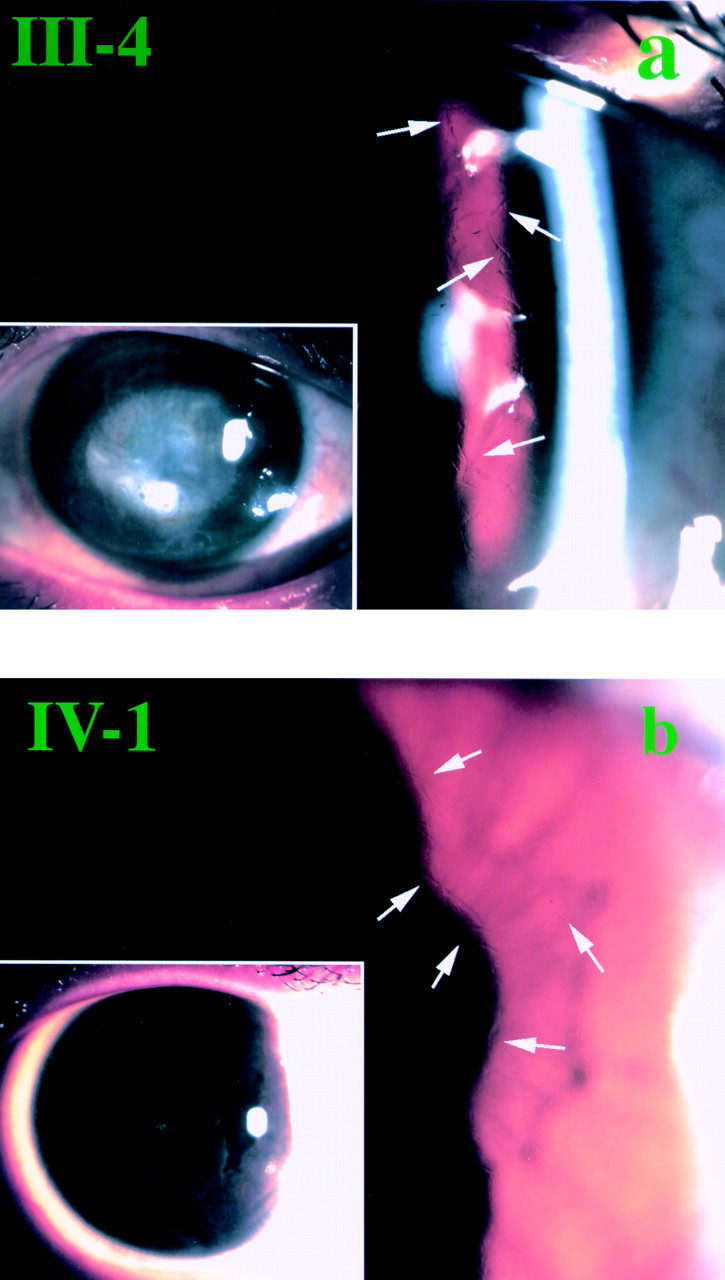
Slit lamp biomicroscopic findings. (a) The proband (III-4). Refractile lattice lines (arrows) are seen in the mid-peripheral area of the cornea. Inset shows the central corneal opacity. (b) Patient IV-1. Subepithelial white dots and placoid pattern of corneal opacity are seen in the inset. Refractile fine lines (arrows) can be seen.
A deep lamellar keratoplasty (DLK) was performed on his left eye (by HK), and his visual acuity improved to 20/60 NC 10 days after surgery.
Patient IV-1 is a 17 year old girl who first visited an ophthalmologist because of ocular pain at 14 years of age and was followed by the same doctor. Her visual acuity gradually decreased and on her first visit to the Nagoya University Hospital, it was 20/400 (20/30 × −5.0D=cyl −1.50D Ax 180) right eye, and 20/300 (20/100 × −3.5D=cyl −3.25D Ax 180) left eye. Her IOPs were 14 mm Hg right eye and 10 mm Hg left eye. Fine lattice lines were observed in the superficial and midstromal area of the corneas of both eyes. The central corneal opacity was not as dense as that of the proband, and instead, fine white dots and faint placoid pattern of superficial opacity were observed (Fig 2b). By a careful history taking, it was learned that she was a cousin of the proband. Blood examination did not show signs of renal failure or anaemia; however, hypertriglycaemia (280 mg/dl) was detected as it was in the proband (177 mg/dl).
Methods
After obtaining informed consent, molecular genetic analysis was performed. DNA was extracted from peripheral leucocytes in three affected members (III-2, III-4, and IV-1) and one unaffected family member (III-7). Exon 4 of the βig-h3 gene was amplified by polymerase chain reaction (PCR) and directly sequenced.
Histopathological study was performed on the corneal tissue of III-4 obtained during DLK.
Results
All three affected individuals showed a heterozygous single base pair transition (CTG to CCG, leucine to proline) (Fig 3, III-2, III-4, and IV-1). No mutation was detected in Arg124. The unaffected patient did not show any mutation in Leu518 (Fig 3, III-7). The nucleotide sequences were confirmed from both directions.
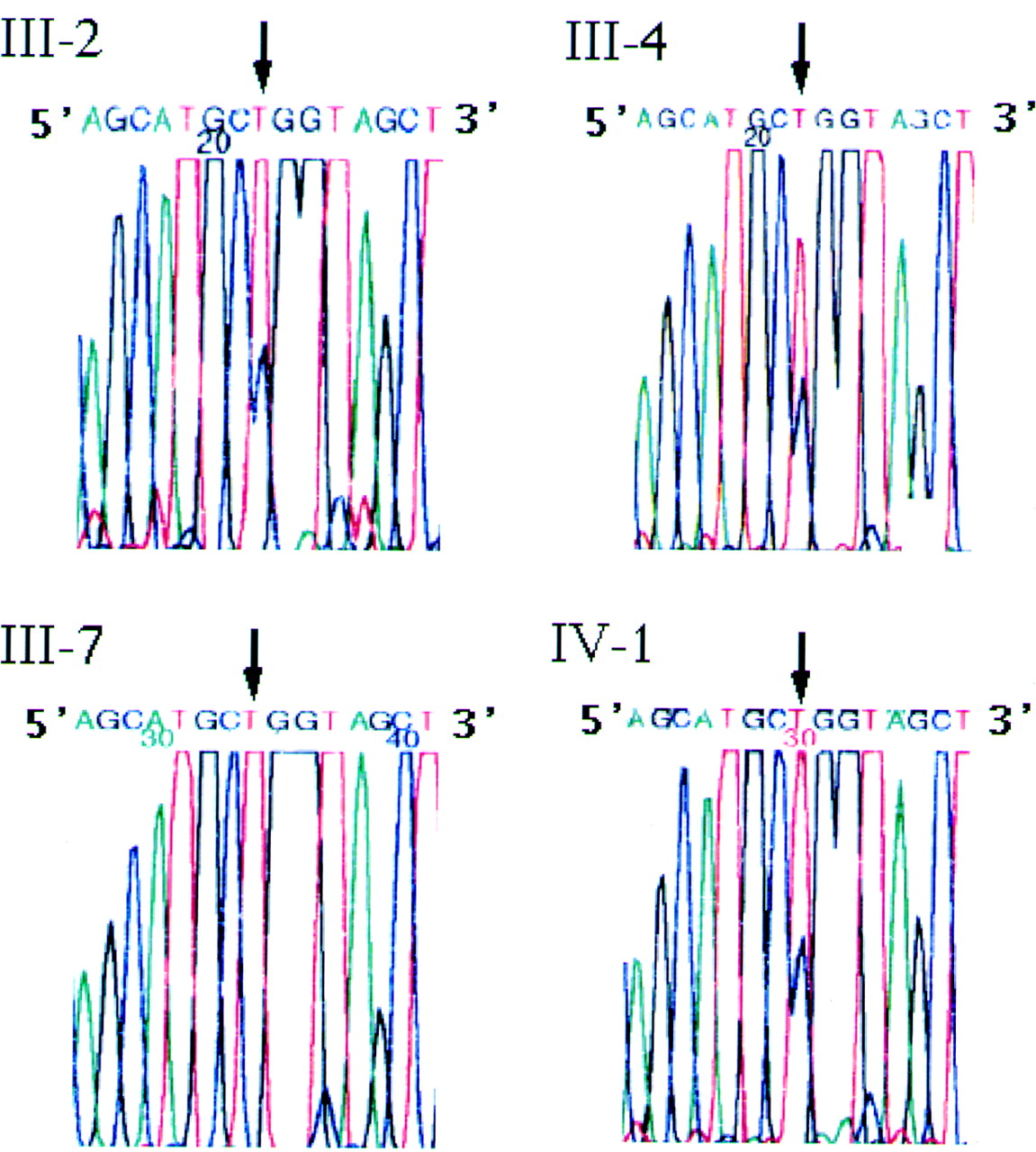
Nucleotide sequences of the βig-h3 gene using sense primer. Three affected individuals (III-2, III-4, IV-1) show a heterozygous single base pair transition (CTG to CCG, Leu to Pro). The proband's unaffected sister (III-7) shows wild type allele.
Histopathological observation of the removed corneal tissue showed that the epithelium varied in thickness and Bowman's layer was interrupted (Fig 4). Amyloid deposits, which were stained pink to orange by congo red, were observed between the collagen bundles and extended into the deep stroma (Fig 4a). The deposits were also observed in the subepithelial area of the epithelium. These deposits demonstrated green birefringence with a polarising filter (Fig 4b).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Histological appearance of the proband's cornea stained with congo red. (a) The epithelium varies in thickness and the interruption of Bowman's layer can be seen (arrowheads). Pink to orange coloured amyloid deposits (arrows) are seen among the bundles of the collagen fibrils. These deposits demonstrate green birefringence with a polarising filter (b, arrows) (bar =100 μm).
Discussion
Two types of corneal amyloidosis, lattice corneal dystrophy type I (LCDI) and Avellino corneal dystrophy, are known to be caused by the R124 mutation of βig-h3 gene, and R124 seems to be responsible for the precipitation of amyloidogenic intermediates in the corneal stroma,6 even though the codon 124 is critical in the pathogenesis of a wide range of corneal dystrophies (Table1).9 10 In a recent report, however, a P501T mutation of βig-h3 was detected in lattice corneal dystrophy type IIIA11 and a L527R mutation in another type of lattice corneal dystrophy with deep stromal opacities,12 instead of a R124 mutation. Table 1 lists the corneal dystrophies caused by the mutation of βig-h3 gene.6-13
Phenotypes of corneal dystrophies caused by the βig-h3 mutations
In the family studied in this report, a L518P mutation instead of an R124 mutation of βig-h3 was detected in the patients with LCDI, and the unaffected member of this family revealed no mutation in the codon 518. These results, together with those from a previous study,8 indicate that the L518P mutation of the βig-h3 gene forms amyloidogeneic intermediates that precipitate in the cornea. By careful family history taking, we learned that the family had no relatives either in Tokyo or in Joetsu where another L518 mutated LCDI family8 reported previously are from. It was thus considered that there is no connection between the two L518 mutated LCDI families.
The clinical features, obtained by the history taking from patients II-6, III-2 and III-5 as well as the two cases described, resemble quite closely those of R124C mutated LCDI. The ocular pain due to epithelial disorders which occurs in their teens is the first symptom, followed by the visual disturbance. Findings with the slit lamp were also identical to R124C mutated LCDI.
This histopathological study is the first conducted on the Leu518Pro mutated lattice dystrophic cornea, and together with the findings in R124 mutated LCDI,7 14 the congo red positive deposits were also detected in this dystrophy. The surgically removed corneal specimens of the proband showed an epithelium of variable thickness, an interrupted Bowman's layer, and amyloid deposits between the collagen bundles of the stroma. These findings are similar to those of LCDI reported earlier.1 2 14 However, these deposits were distributed much deeper in the stroma compared with the R124C mutated LCDI.7
Although the cause of the proband's renal failure is still unknown, hypertriglyceridaemia was found in both the proband and patient IV-1, and abnormal liver function due to fatty liver was detected in III-5. We are searching for the possibility of a general disorder derived from Leu518Pro mutation of βig-h3.
Acknowledgments
Supported by a grant in aid for scientific research from the Ministry of Education, Japan (K Hirano: (C) 11671732, A Kanai: (B) 05454477, (B) 0745741).
We thank Professor Yozo Miyake for invaluable suggestions during the preparation of the manuscript