Article Text

Abstract
AIMS A number of genetic loci have been implicated in the pathogenesis of primary open angle glaucoma (POAG). The aim of this study was to identify the genetic cause of POAG in a large Scottish family and, if possible, offer genetic screening and advice to family members.
METHODS Family members were examined to determine their disease status. Base excision sequence scanning was carried out in order to test for the presence of a POAG causing mutation at known genetic loci. Direct DNA sequencing was performed in order to determine the mutation sequence.
RESULTS All family members of known affected disease status and two family members of unknown disease status were found to have a mutation in theTIGR gene. The mutation resulted in the substitution of a glycine residue with an arginine residue at codon 252 (Gly252Arg). No other sequence variations were present in any members of the family.
CONCLUSION The Gly252Arg mutation in the TIGR gene results in the development of POAG in this family. It was possible to identify younger, currently unaffected, members of the family who carry the mutation and who are therefore at a very high risk of developing POAG themselves. This is the first demonstration that Gly252Arg can be a disease causing mutation rather than a benign polymorphism. The possible pathogenic mechanisms and wider implications of the mutation are considered.
- primary open angle glaucoma
- genetic screening.
Statistics from Altmetric.com
Genetic factors play an important part in the aetiology of primary open angle glaucoma (POAG).1 It has long been recognised that relatives of patients with POAG have an increased risk of developing the condition themselves compared with the general population. Estimates of the increased prevalence range from 2.8% to 13.5%.2 Twin studies have shown a degree of concordance consistent with a polygenic or multifactorial inheritance for POAG.3 Most POAG families do not show a simple Mendelian pattern of inheritance. Nevertheless, there have been a number of POAG families described in which the disease shows an autosomal dominant form of inheritance.4-8 Genetic analysis of such families has implicated a number of regions of the human genome in the pathogenesis of POAG, as shown in Table 1.9-14 The only causative gene cloned from any of these loci at present is the trabecular meshwork inducible glucocorticoid response gene (TIGR).15 Mutations in this gene are responsible for glaucoma in those families which show linkage to the GLC1A region on chromosome 1.15
Human loci implicated in the pathogenesis of primary open angle glaucoma
The aim of this study was to identify the genetic cause of POAG in a large Scottish family and, if possible, offer genetic screening and advice to family members.
Materials and methods
CLINICAL EXAMINATION
Every member of the family was examined by one of the authors (AB). Examination consisted of applanation tonometry, slit lamp biomicroscopy, gonioscopy, funduscopy, and perimetry. Subjects were labelled as POAG affected if they had an open anterior chamber angle, glaucomatous optic disc damage, and glaucomatous visual field loss. Raised intraocular pressure (IOP) was not necessary for the diagnosis of POAG in order not to exclude potential subjects with normal tension glaucoma. Subjects were labelled as unaffected if there was no evidence of optic disc damage or glaucomatous field loss and the individual was aged 35 or older. Subjects were labelled as ocular hypertension if they had an IOP above 21 mm Hg in the absence of glaucomatous optic disc or field changes. All other individuals were considered to be of unknown disease status.
Blood samples from all family members were collected into tubes containing EDTA in order to allow subsequent DNA extraction.
The study was approved by the appropriate local medical ethics committees.
POLYMERASE CHAIN REACTION (PCR)
Genomic DNA was extracted from blood using the Nucleon genomic DNA kit according to the manufacturer's protocol. PCR was performed in a Techne thermal cycler PHC-3 using genomic DNA (5 ng/μl), sequencing grade Taq DNA polymerase (Promega), and PCR primers (10 pmol/μl) (Oswell DNA Service), as detailed in Table 2. PCR products were purified using the QIAquick PCR purification kit (Qiagen).
Oligonucleotide primers used in the study
BASE EXCISION SEQUENCE SCANNING (BESS)
BESS was performed using labelled primers as shown in Table 2. BESS samples were prepared using the BESS T-scan mutation detection and localisation kit (Epicentre Technologies). Samples were electrophoresed, detected, and analysed using a Li-Cor DNA Sequencer Model 4000L and Base ImageIR 4.0 software (Li-Cor).
DNA SEQUENCING
Direct dideoxy sequencing was performed on purified PCR amplified fragments using 32P end labelled primers.16Reactions were carried out using the T7 Sequenase version 2.0 DNA sequencing kit (Amersham Life Science). Sequencing reaction products were resolved on a buffer gradient (0.5–2.5X TBE) 40% Acrylamide/Bis (19:1) (BioRad) sequencing gel. DNA sequencing bands were detected by autoradiography.
Results
CLINICAL EXAMINATION
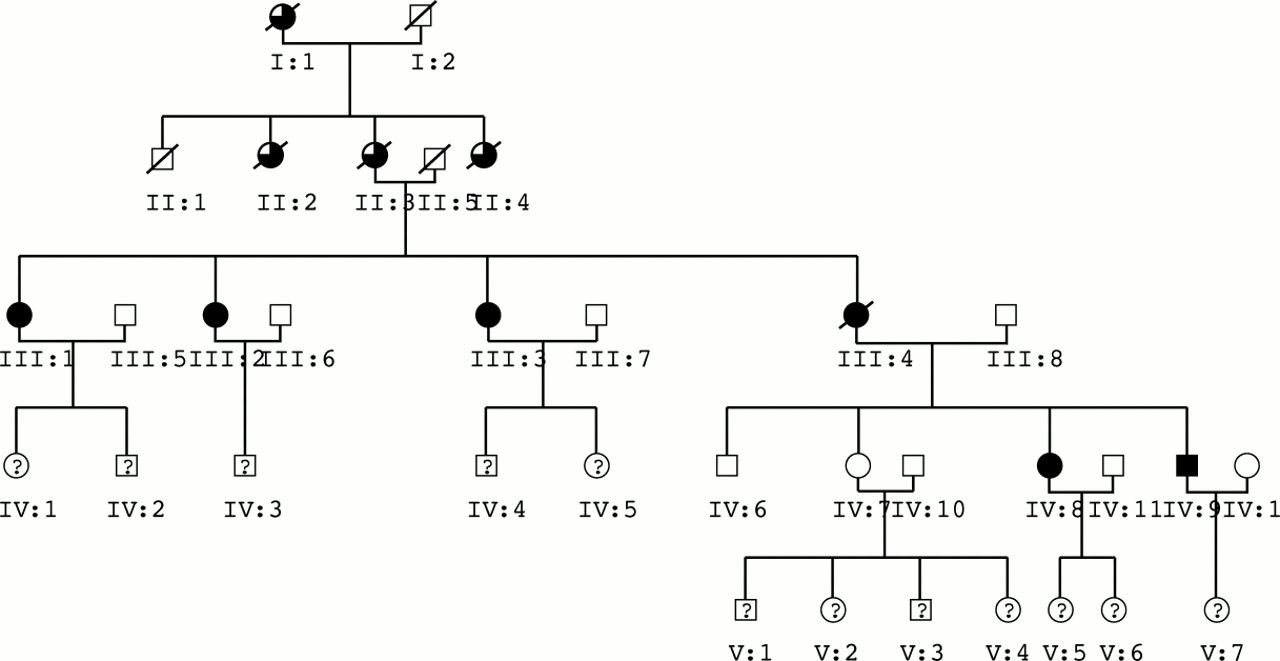
The study family comprises five generations and exhibits an autosomal dominant pattern of inheritance (Fig 1). DNA samples were obtained from most living members of the family. The clinical features of affected individuals are shown in Table 3. The ages of onset of glaucoma are approximate for individuals III:1, III:2, III:3, and III:4 because, at the time of diagnosis, all showed signs of relatively advanced glaucoma. It is likely therefore that they had all suffered glaucoma for an indeterminate period of time before diagnosis. However, as a result of the recognition of the strong family history of glaucoma, individuals IV:8 and IV:9 underwent regular ophthalmic examinations, thereby allowing the condition to be diagnosed at an early stage.

Family tree. Solid symbols indicate affected disease status. Open symbols indicate unaffected disease status. Part solid symbols indicate deceased family members with history suggestive of POAG. Question marks indicate unknown disease status in living family members.
Clinical features of affected members of the study family
All affected family members demonstrated a raised intraocular pressure greater than 21 mm Hg at presentation, with variable glaucomatous optic disc and field changes. No individuals were diagnosed as having ocular hypertension—that is, all unaffected family members had an IOP less than 22 mm Hg. All family members had an open anterior chamber angle on gonioscopy. All affected individuals, however, showed the presence of abnormal tissues in the anterior chamber angle, as detailed in Table 3.
MUTATION ANALYSIS
All affected individuals developed their glaucoma by the fourth decade. This therefore suggested a juvenile onset type of POAG. Juvenile onset POAG (J-POAG) has previously been linked to theGLC1A locus.9 Initial mutation analysis was therefore focused on the TIGRgene, which has been shown to be mutated inGLC1A linked glaucoma.15
The TIGR gene was amplified by PCR using the primers detailed in Table 2. BESS analysis revealed an extra band in PCR products amplified with the primer pair TIGRIN2F/TIGR5R in all known affected individuals (Fig 2). An extra band was also detected in samples from individuals IV:2 and V:5, who were of unknown affectation status. This suggested the presence of a mutation in theTIGR gene at a point between the two PCR primers TIGRIN2F and TIGR5R.

BESS analysis of the TIGR gene between primers TIGRIN2F and TIGR5R. An extra band is seen in all individuals of known affected disease status III:1, III:2, III:3, IV:8, and IV:9. An extra band is also seen in individuals IV:2 and V:5 of unknown disease status (marked with an asterisk).
Direct sequence analysis revealed a heterozygous change at base pair 790, from guanine to adenine (G790A), as shown in Figure 3. The base change was confirmed by sequencing of the reverse strand. This base pair change results in the substitution of the amino acid glycine by the amino acid arginine at codon 252 (Gly252Arg). No other sequence variations were detected in any family members.

{kind=link}
{kind=link}
{kind=link}
Sequence analysis of the TIGR gene. Codon 252, containing the mutation in affected individuals, is highlighted. The normal sequence, GGA (glycine), is seen in individual IV:6 (unaffected disease status). The mutated sequence, AGA (arginine), is seen in individual IV:8 (affected disease status). This results in an amino acid change from glycine to arginine at codon 252 (Gly252Arg).
The Gly252Arg mutation co-segregates with POAG in this family, as shown by the BESS results in Figure 2. The Gly252Arg mutation is thus the likely cause of POAG in the affected family members. In addition, individuals IV:2 and V:5 also harbour the same mutation in theTIGR gene. They are therefore at a high risk of developing J-POAG.
Discussion
CLINICAL
The family described in this study consists of five generations with a very strong history of POAG. All affected individuals developed the condition by their fourth decade. More exact ages of disease onset are not known as a result of relatively advanced glaucoma in a number of individuals at the time of first presentation. The glaucoma in this pedigree was particularly severe, with five out of the six clinically affected individuals requiring bilateral trabeculectomies. The indication for trabeculectomy was lack of medical control of IOP and/or progressive field loss. This is in keeping with the previously reported aggressive nature of J-POAG.17
All affected individuals on whom gonioscopy was performed had open drainage angles. Nevertheless, they all also had either abnormal blood vessels and/or mesodermal tissue present in the drainage angle. The majority of J-POAG pedigrees linked toGLC1A have not had any abnormalities observed on gonioscopy.4 17-19 However some J-POAG pedigrees, linked to GLC1A, have been described with signs of goniodysgenesis or iris processes.20 21 Reflecting this apparent variation in anterior chamber angle appearance in J-POAG, a recent study found that in 231 patients with juvenile glaucoma, 8% had a closed angle and 92% an open angle.22 Of the patients with an open angle, 57% showed no gross pathological features on gonioscopy while 35% showed signs of goniodysgenesis. It has also been suggested that the trabecular meshwork is smaller in eyes with J-POAG compared with normal eyes.23
GENETIC
All known affected family members carry the Gly252Arg mutation. In addition, two individuals of previously unknown affectation status harbour the same mutation.
The gene sequence variation G790A has previously been described in a family with POAG.24 The clinical picture was very similar to our family. However, as the previously described family contained only one affected individual, it was unclear as to whether the resultant Gly252Arg amino acid change was a disease causing mutation or a benign polymorphism.25 This is because, with only one affected member in the family, it was impossible to demonstrate co-segregation of the disease with the mutation. In this family, however, it is possible to trace co-segregation of the disease with the mutation. This strongly suggests therefore that Gly252Arg is a disease causing mutation rather than a benign polymorphism. In addition, Rozsa and colleagues have previously shown that a Gly252Arg substitution was not present in 43 normal individuals aged 35–83 years (mean 55 years), thus suggesting, but not confirming, that the Gly252Arg substitution was more likely to be a mutation than a polymorphism.25
Mutations in the TIGR gene are estimated to account for 10% of familial cases of both J-POAG and A-POAG, as well as 5% of sporadic mutations.26-28 TheTIGR gene consists of three exons (coding regions) separated by two introns (non-coding regions).29In addition, the gene has a large 5 kb promoter region which controls expression of the gene.30-31 TIGR has been accurately mapped by fluorescent in situ hybridisation to chromosome 1q23-q24.32 The gene encodes a protein of 504 amino acids in length.30 The C terminal end of the TIGR protein, from codons 324–502, shows a strong sequence homology (sequence similarity) to a protein found in the neuro-olfactory epithelium of the nose, called olfactomedin.29 33 The vast majority ofTIGR mutations associated with POAG lie within this region of olfactomedin homology. This suggests that the region of olfactomedin homology plays an important, though as yet unknown, part in the pathogenesis of POAG. Olfactomedins are known to oligomerise via disulphide bond formation, a property which is also thought to occur with the TIGR protein. Mutations within this region could therefore potentially result in the abnormal accumulation of TIGR protein. This might restrict the exit of aqueous humour through the trabecular meshwork, leading to the raised IOP commonly seen in POAG. The Gly252Arg mutation is therefore unusual as it lies outside this region of high mutation density. The mutation also lies outside a leucine zipper region (codons 117 to 166) which has been implicated as another potential site for TIGR dimerisation and oligomerisation.30
Glycine has a small uncharged side chain, while arginine has a large positively charged basic side chain; the Gly252Arg mutation therefore alters the local charge density. In addition, the Chou-Fasman algorithm predicts that the Gly252Arg mutation causes a change in protein secondary structure from a β strand to an α helix between residues 245 and 264.25 This results in an alteration of the structure around both a potentially important protein kinase C phosphorylation site (residues 256–258, Thr-Leu-Arg) and a highly conserved cysteine residue (residue 245).
There is therefore very strong evidence that Gly252Arg is a mutation rather than a polymorphism. Firstly, it shows co-segregation with the disease in our family. Gly252Arg was previously shown not to be present in 43 normal individuals.25 Finally, it results in a significant alteration in both the local charge density and protein structure of TIGR.
GENETIC SCREENING
Neither individual IV:2 nor individual V:5 showed any signs of glaucoma at the time of their examination for this study, when they were aged 16 and 26 respectively. However, sequence analysis shows that both individuals carry the Gly252Arg mutation. The penetrance of this Gly252Arg mutation is not known. The penetrance of two otherTIGR mutations has however, been calculated as 90% for the Lys423Glu mutation and >95% for the Asn480Lys mutation.34 35 If the Gly252Arg mutation shows a similar rate of penetrance, then individuals IV:2 and V:5 are at a very high risk of developing the disease. It will therefore be important to monitor them closely in order to detect any early sign of the development of POAG.
A more complete understanding of the prevalence of the differentTIGR mutations may allow screening to be made available to a wider, more general, population rather than be restricted to a few selected families. Such a screening programme, however, would need to take into account the effects of the other loci implicated in the aetiology of POAG, as detailed in Table 1. A negative screening test for a TIGR mutation does not exclude the possibility of POAG developing from these other genetic aetiologies. In the future, our improved understanding ofTIGR and its role in the pathogenesis of glaucoma may allow the development of new, innovative therapeutic products. More distantly, there is the possibility of gene therapy for those people who carry a mutation in the TIGRgene.
In summary, our study demonstrates that a Gly252Arg substitution in theTIGR gene is likely to be a pathogenic mutation rather than a benign polymorphism. In addition, we have demonstrated the clinical value of genetic screening of families with a strong history of POAG. It is hoped that, in the near future, genetic screening for POAG can be made available to a wider population.
Acknowledgments
We would like to thank all members of the family for their enthusiastic support of this study. We would also like to thank all the ophthalmologists who allowed their patients to be included in this study. This work was supported by the Northern and Yorkshire Regional Health Authority, the Medical Research Council, the International Glaucoma Association, the Glaucoma Research Foundation, That Man May See, and Insite Vision.