Article Text

Abstract
Aim: To study the normal distributions of mitochondria and voltage gated Na+ channels in the human optic nerve head in order to gain insight into the potential mechanisms of optic nerve dysfunction seen in the inherited optic neuropathies.
Methods: Five fresh frozen human optic nerves were studied. Longitudinally orientated, serial cryosections of optic nerve head were cut for mitochondrial enzyme histochemistry and immunolabelling for cytochrome c oxidase (COX) subunits and voltage gated Na+ channel subtypes (Nav 1.1, 1.2, 1.3, and 1.6).
Results: A high density of voltage gated Na+ channels (subtypes Nav 1.1, 1.3, and 1.6) in the unmyelinated, prelaminar, and laminar optic nerve was found. This distribution co-localised both with areas of high COX activity and strong immunolabelling for COX subunits I and IV.
Conclusions: Increased numbers of mitochondria in the prelaminar optic nerve have previously been interpreted as indicating a mechanical hold up of axoplasmic flow at the lamina cribrosa. These results suggest that this increased mitochondrial density serves the higher energy requirements for electrical conduction in unmyelinated axons in the prelaminar and laminar optic nerve and is not a reflection of any mechanical restriction. This could explain why optic neuropathies typically occur in primary inherited mitochondrial diseases such as Leber’s hereditary optic neuropathy, myoclonic epilepsy with ragged red fibres (MERRF), and Leigh’s syndrome. Secondary mitochondrial dysfunction has also been reported in dominant optic atrophy, Friedreich’s ataxia, tobacco alcohol amblyopia, Cuban epidemic optic neuropathy, and chloramphenicol optic neuropathy. These diseases are rare but these findings challenge the traditional theories of optic nerve structure and function and may suggest an alternative approach to the study of commoner optic neuropathies such as glaucoma.
- mitochondria
- myelination
- optic nerve
- sodium channels
Statistics from Altmetric.com
The optic nerve is an extension of the central nervous system and has some unique structural features which potentially make it vulnerable to disease processes. At an ultrastructural level unmyelinated nerve fibres exit the eye via the lamina cribrosa becoming myelinated at its posterior border. On electron microscopy, mitochondria are concentrated in the prelaminar and laminar regions1,2 and in our previous studies in pig, rabbit, and normal human optic nerve we demonstrated histochemically and immunohistochemically that mitochondrial enzyme activity and immunoreactivity is markedly higher in these unmyelinated regions.3,4 This distribution of mitochondria has traditionally been attributed to mechanical constriction or axoplasmic stasis at the lamina.2 We suggest that mitochondrial distribution has little to do with lamina structure but rather reflects the energy requirements to maintain conduction in unmyelinated compared to myelinated axolemma. If this hypothesis is correct we would expect the machinery that generates and propagates action potentials—that is, axonal voltage gated Na+ channels, to co-localise with mitochondrial activity. In this work we studied human optic nerve investigating the distribution of mitochondria and voltage gated Na+ channel subtypes in relation to myelination and the position of the lamina cribrosa.
MATERIALS AND METHODS
Collection and preparation of ocular tissue
We collected eyes from deceased donors following the harvesting of corneoscleral discs for transplantation. Globes were rapidly frozen in isopentane, previously cooled in liquid nitrogen, and stored at −80°C. Blocks of optic nerve were later dissected from the frozen globe using a pathologist’s disposable trimming blade. The resulting block was then mounted in cryoembedding matrix (“Cryo-M-Bed” Bright Instrument Co, Huntingdon, UK) and 10 µm, longitudinal sections cut in a cryostat (CryoStar HM560M: Microm International, Walldörff, Germany). All sections were air dried for 1 hour at room temperature before storage in airtight containers at −80°C.
Histological and enzyme histochemical techniques
Haematoxylin and eosin, Van Gieson’s, and Weigert’s iron haematoxylin stains were employed according to standard methods.5
Cytochrome c oxidase (COX) activity was demonstrated by incubating sections in a solution containing 100 µM cytochrome c, 4 mM 3,3′-diaminobenzidine tetrahydrochloride, 20 µg/ml catalase in 0.2 M phosphate buffer, pH 7.0, at 37°C for 45 minutes.6 The sections were then washed in phosphate buffered saline (PBS), dehydrated in a graded ethanol series (70%, 95%, 2×100%), cleared in Histoclear (National Diagnostics, Atlanta, GA, USA), and mounted in a synthetic resin (DPX, BDH, Poole, UK).
Primary antibodies
Voltage gated sodium channels
-
Na(v) 1.1 (diluted 1:100) and Na(v) 1.2 (diluted 1:50); goat polyclonals (Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA)
-
Na(v) 1.3 (diluted 1:100); rabbit polyclonal (Alomone Laboratories, Jerusalem, Israel)
-
Na(v) 1.6 (diluted 1:50); rabbit polyclonal (Europa Bioproducts Ltd, Ely, UK).
COX subunits
-
COX-I (diluted 1:500) and COX-IV (diluted 1:200); mouse monoclonals (Molecular Probes, Eugene, OR, USA)
-
Neurofilament protein (200 kD) (diluted 1:200); rabbit polyclonal (Chemicon International, Temecula, CA, USA).
Immunohistochemistry
Voltage gated sodium channels and neurofilament protein
Sections for voltage gated Na+ channel and neurofilament protein immunohistochemistry (IHC) were allowed to reach room temperature and air dried for a further hour. The sections were then placed in PBS, containing 0.1% (v/v) Triton X-100 (PBST) for 15 minutes. Primary antibody (diluted in 4% (w/v) bovine albumin (fraction V, Sigma, Poole, UK) in PBST) was then applied for 1 hour at room temperature. Following three 5 minute washes in PBST endogenous peroxidase activity was quenched by treatment with 0.3% (v/v) hydrogen peroxide in methanol for 15 minutes. A 5 minute wash in PBST was then followed by incubation in the appropriate, species specific (anti-goat, anti-rabbit (Dako Ltd, Ely, UK)), peroxidase conjugated secondary antibody (diluted 1:100; diluent as above) for 1 hour at room temperature. Sections were then washed in PBST as before and peroxidase activity demonstrated (5 minute incubation) using a commercial kit (Vector Laboratories, Burlinghame, CA, USA) employing 3, 3′-diaminobenzidine tetrahydrochoride as chromogen.
A wash in distilled water was followed by counterstaining in Mayer’s haematoxylin. Sections were then dehydrated, cleared and mounted as above.
COX subunits
Although IHC for COX subunits was similar to that for voltage gated Na+ channels, a number of procedures differed; following air drying the sections were fixed in 4% (w/v) paraformaldehyde in 0.1 M phosphate buffer, pH 7.4, for 10 minutes at 4°C. The permeabilisation step in PBST used above was followed by an additional permeabilisation step combined with a step to quench endogenous peroxidase activity thus; 70% (v/v) methanol, 10 minutes, 95% methanol (v/v) plus 0.3% (v/v) hydrogen peroxide, 10 minutes, 100% methanol, 20 minutes, 95% (v/v) methanol plus 0.3% hydrogen peroxide (v/v), 10 minutes, 70% (v/v) methanol, 10 minutes, and finally PBST, 5 minutes.
Microscopy and image capture
Sections were examined and images captured using a Zeiss Imaging System consisting of an Axioplan 2iE microscope, AxioCam HRc digital camera and AxioVision image capture software (Imaging Associates, Bicester, UK).
RESULTS
We investigated optic nerves from five people (table 1). Haematoxylin and eosin staining was used to assess the preservation of morphological integrity (fig 1A) while Van Gieson’s stain was used to demonstrate the position of the lamina cribrosa (fig 1B). Weigert’s iron haematoxylin confirmed the abrupt initiation of myelination in the retrolaminar portion of the optic nerve (fig 1C). COX activity (fig 1D) was undetectable in the myelinated optic nerve but was high in prelaminar and intralaminar regions where myelin is absent. To strengthen the results of the COX histochemistry, IHC for COX I and IV subunits was used. An identical immunolabelling pattern to that for COX activity was observed for both subunits (COX I, not shown). Only very pale immunolabelling of the myelinated portion of the optic nerve was seen, particularly with the COX IV subunit antibody, but this was barely distinguishable from background (fig 1E).
Patient details

Histology, enzyme histochemistry and immunohistochemistry on 10 µm, serial, cryostat sections of optic nerve from a 45 year old woman. (A) Haematoxylin and eosin preparation showing preservation of morphology. (B) Van Gieson preparation to demonstrate connective tissue fibres (red). The position of the lamina cribrosa is indicated by the arrow. (C) Weigert’s iron haematoxylin preparation to demonstrate myelin (dark blue). Note that myelination commences abruptly in the retrolaminar position. (D) COX enzyme histochemistry showing strong reaction in the unmyelinated portion of the optic nerve extending into the lamina cribrosa but halting abruptly in the retrolaminar position. (E) IHC for COX subunit IV showing that the distribution of the protein co-localises with enzyme activity. (F) IHC for Nav 1.1 showing that the distribution of this voltage gated Na+ channel parallels that of the mitochondrial markers. (G) IHC for Nav 1.2 showing a very pale, diffuse labelling pattern with no predilection for either prelaminar or retrolaminar positions. (H) IHC for Nav 1.6 showing strong immunolabelling in unmyelinated optic nerve. (I) Representative negative control for IHC where the primary antibody was omitted.
IHC for CNS associated voltage gated Na+ channels showed distributions identical to that revealed by the mitochondrial markers. Nav 1.1 showed marked immunolabelling in unmyelinated optic nerve while in the retrolaminar, myelinated portion, immunolabelling was undetectable (fig 1F). Nav 1.3 and Nav 1.6 (fig 1G) showed very strong immunolabelling with an identical distribution as that seen for Nav 1.1. By contrast, Nav 1.2 showed very pale and diffuse immunolabelling which was not confined to any particular region of the optic nerve (fig 1F). Omission of the primary antibody excluded the possibility of non-specific immunolabelling by the secondary antibody and allowed the effectiveness of endogenous peroxidase quenching to be assessed (fig 1I).
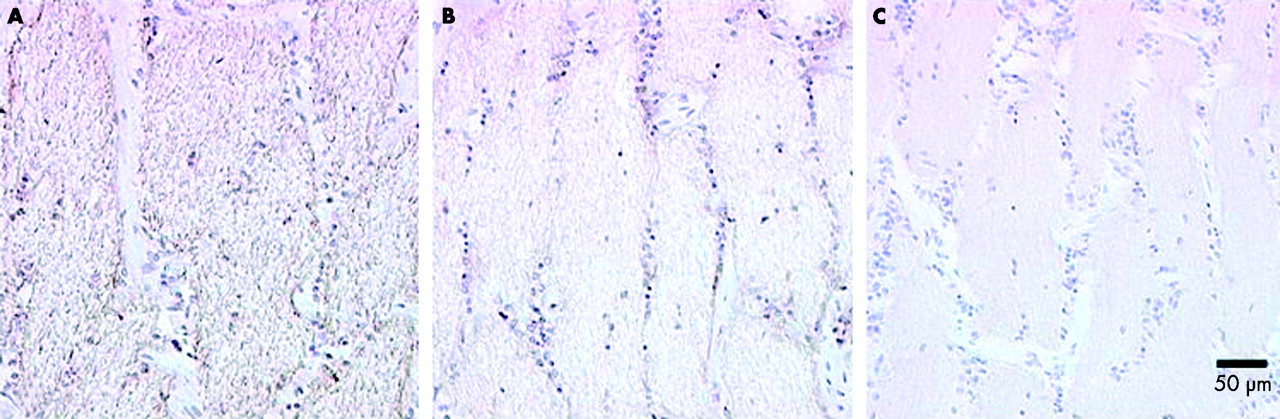
To address concerns regarding the possible exclusion of immunoreagents from the myelinated portion of the optic nerve we compared the immunolabelling produced with the COX IV subunit antibody, Nav 1.6 antibody, and an antibody to neurofilament protein (200 kDa), an axonal marker. The myelinated portion of the optic nerve showed a strong, fibrous immunolabelling pattern with the neurofilament antibody. Very pale immunolabelling was detected with the Nav 1.6 and COX IV antibodies (fig 2). Thus it would appear immunoreagents have full access to myelinated axons using our methodology.

{kind=link}
{kind=link}
Control experiment to show immunoreagents have access to the myelinated portion of the optic nerve. (A) IHC for neurofilament protein (200 kDa) showing a strong, fibrous immunolabelling pattern in myelinated optic nerve. (B) IHC for Nav 1.6. (C) COX subunit IV, showing pale immunolabelling.
DISCUSSION
The optic nerve head extends from the optic disc to the posterior scleral surface. Within that short distance (0.5 mm) the ganglion cell axons traverse the lamina cribrosa and become organised into bundles. Myelination begins at the posterior edge of the lamina cribrosa.
The cell bodies of the ganglion cell axons lie within the retina and a transport system exists to move proteins to the distal axon by anterograde flow.7 The concept of a mechanical hold up of axoplasmic flow at the lamina cribrosa followed the observation of an accumulation of organelles, principally mitochondria, at the laminar and prelaminar optic nerve head.2 It has been proposed that the transition from the relatively high pressure intraocular environment to the lower orbital and intracranial environment results in mechanical constriction of ganglion cell axons at the level of the lamina cribrosa. It has also been suggested that in glaucoma raised intraocular pressure results in an exaggeration of this normal chronic obstruction to axoplasmic flow ultimately resulting in cell death.8
An alternative explanation for the increased numbers of mitochondria seen at the laminar and prelaminar optic nerve is that they serve the high energy requirements for conduction in unmyelinated axons. Myelinated axons conduct more efficiently by saltatory conduction, with depolarisation and repolarisation only occurring at the nodes of Ranvier.9,10 In human optic nerve the junction between unmyelinated and myelinated axolemma is at the posterior border of the lamina cribrosa. Previous studies, in rabbit, where myelination extends on to the retina, have shown that the change in mitochondrial activity is displaced on to the retinal surface to the point where myelination commences.4 This is further evidence that it is the presence or absence of myelin that determines the density of mitochondria in an axon rather than mechanical factors at the lamina.
Our results show that voltage gated Na+ channel subtypes Nav 1.1, 1.3, and 1.6 are found at much higher density in the prelaminar and laminar optic nerve and that the density declines dramatically with the onset of myelination. This implies that the distribution of mitochondria in the optic nerve head reflects functional energy requirements to maintain conduction in the unmyelinated axon rather than any mechanical restriction at the lamina cribrosa under normal physiological conditions. We accept that mechanical hold up might occur under condition of raised intraocular pressure but if it does it is not an exaggeration of a normal physiological process.2
A previous report of voltage gated Na+ channel distribution in rat optic nerve showed Nav 1.2 localisation within the unmyelinated portion and that of Nav 1.6 confined to the nodes of Ranvier.11 However, species differences in voltage gated Na+ channel expression are not unprecedented. For example, Nav 1.3 is very restricted in adult rat central nervous system12 but is strongly expressed in adult human brain.13 In the latter study, immunolabelling for Nav 1.1, 1.3, and 1.6 was largely limited to neuronal cell bodies and their proximal processes, whereas Nav 1.2 was confined to axons proper. If that portion of the axon between the axon hillock and the commencement of myelination is considered to be a proximal process then the equivalent in the optic nerve would be the prelaminar and intralaminar region. Thus the unmyelinated portion of the optic nerve may be homologous to proximal processes in general and share the same distribution of voltage gated Na+ channel subtypes.
It is already known that a number of primary inherited disorders of mitochondrial dysfunction target the optic nerve. These include Leber’s hereditary optic neuropathy, Leigh’s syndrome, and myoclonic epilepsy with ragged red fibres (MERRF). In addition, disorders of secondary mitochondrial dysfunction also target the optic nerve. These include Friedreich’s ataxia, dominant optic atrophy, tobacco alcohol amblyopia, Cuban epidemic optic neuropathy, and cheoramphenicol optic neuropathy.14
Of particular interest in this regard is dominant optic atrophy caused by mutations in the OPA1 gene.15 This encodes a dynamin related GTPase which is involved in mitochondrial interaction with the cytoskeleton.16,17 Experimental studies have shown mitochondrial clumping as a consequence of the mutant protein15 and OPA1 absence has recently been shown to induce apoptosis.18 This suggests that mutations of the OPA1 gene may affect the physiological distribution of mitochondria within the optic nerve and lead to impaired cell viability.
These diseases are rare but our findings challenge the traditional theory of a mechanical hold up of axoplasmic flow at the lamina in glaucoma by providing a better explanation for the increased numbers of organelles (principally mitochondria) observed in the lamina and prelaminar optic nerve head.