Article Text

Abstract
Aims To report observations relating to the clinical recognition and possible basis of reticular pseudodrusen (RPD).
Methods This retrospective study reports the evolution of RPD in 166 patients who had follow-up of over 1 year using multiple imaging techniques. Mean age when first seen was 73.3 years and the mean period of observation was 4.9 years (range 1–18 years). Associated macular changes were recorded.
Results RPD were first identified in the upper fundus as a reticular network, which then became less obvious, developing a diffuse yellowish appearance. RPD also faded around choroidal neovascularisation (CNV). RPD therefore could be transient but the pattern often remained visible outside the macula or nasal to the discs. Manifestations of age-related macular degeneration (AMD) were present in nearly all eyes and there was a particularly high association with CNV (52.1%). In one clinicopathological case abnormal material was found in the subretinal space.
Conclusions The prevalence of RPD may be underestimated because their recognition depends upon the imaging method used, the area of fundus examined and the confusion with typical drusen. The pathology of one eye suggests that RPD may correspond to material in the subretinal space.
- Drusen
- reticular pseudodrusen
- macula
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Introduction
Following the original description in 1990 by Mimoun et al,1 the term pseudodrusen has been used to describe a yellowish interlacing pattern in the fundus of patients with AMD that was most apparent in red-free or blue light but did not show discrete hyperfluorescence on fluorescein angiography (FA). Klein2 referred to them as reticular drusen in the Wisconsin grading system, but recent imaging studies have led to further understanding and consequently to alternative terminologies. Smith et al3 showed that different imaging techniques suggested preferential involvement at varying depths in the retina and choroid and therefore proposed the term ‘reticular macular disease’. Zweifel et al4, using point-to-point correspondence between autofluorescent images and spectral domain optical coherence tomography (OCT), were able to correlate RPD with abnormal material lying internal to the retinal pigment epithelium (RPE) and they therefore favoured the term ‘subretinal drusenoid deposits’. However, there is still no consensus as to the prevalence of RPD or of their relationship to AMD. The present study of the evolution of RPD was undertaken in order to aid in their clinical recognition and to comment further on their possible basis.
Materials and methods
This study reports the findings in 166 patients (32 men and 134 women) who had follow-up of at least 1 year from the time that RPD were first noted (table 1). In 146 patients the RPD were observed at first visit, in 20 patients (31 eyes) the RPD developed during observation. The mean age when RPD were noted was 73.3 years and mean follow-up was 4.9 years (range 1–18 years). In those patients who presented with active CNV the RPD were usually first noted in the second eye, but then could often be identified in both eyes.
Follow-up periods of 166 patients from time RPD first detected
Documentation included colour and red-free fundus photography and, in most eyes, fluorescein angiography with the Zeiss fundus camera or Topcon Imagenet 2000 system. Blue light photographs were obtained using the excitation filter on the fundus camera. Autofluorescence imaging using the Heidelberg Retina Angiograph 2 and time domain or spectral domain OCT using the Zeiss Stratus or Cirrus was performed on selected patients.
The type of associated macular degeneration was recorded and graded according to a simple scale as follows: no drusen or small drusen only, intermediate drusen (63–124 μm) and large drusen over 125 μm. Note was also made of hyperpigmentation, geographic atrophy (GA) if over 0.5 disc in diameter and involving the fovea, adult vitelliform lesions and CNV.
The search of a large collection of eyes previously submitted to clinicopathological examination found one male patient in whom this appearance had been noted but not understood at the time. He had developed small drusen over a 10-year period before he died in 1981 aged 87. On his last visit 7 months before fatality vision had been 6/18 and cataracts blurred the view of his fundi, but RPD had been noted in the right eye. The eye was fixed 3 h 45 min after fatality and prepared for transmission electron microscopy by standard techniques. When re-examined recently, the glutaraldehyde-fixed epoxy embedded blocks were found to be in good condition and fresh semithin (0.5 μm) sections were cut and stained with methylene blue-basic fuchsin. Ultrathin (120 nm) were cut in areas of interest and stained with uranyl acetate and lead citrate.
The study protocol was approved by the University of New South Wales Human Research Ethics Committee number 05115.
Results
The distinction between RPD and typical drusen could be difficult on funduscopy alone, especially where they merged. RPD had a more uniform size and shape, but lacked the thickness of drusen. They did not obscure typical drusen but could be seen to lie anterior to the choroidal vessels and also to naevi. Additional distinguishing features were their distribution, subsequent evolution and imaging characteristics.
Rather than developing individually like typical drusen, RPD were not identified until there were sufficient numbers to establish a reticular pattern (figure 1C,F). RPD were most often first noted between the upper edge of the fovea and the superior temporal vessels but appeared to spare the fovea. RPD could develop relatively suddenly, and over several years spread occurred by continuity (figure 1G), mainly in an upward direction and even into the mid-periphery. To a lesser degree they also developed in other quadrants and they could approach as closely to the edge of the optic disc as the termination of the RPE. The reticular network then became tighter and ultimately disappeared such that the fundus assumed a diffuse yellowish or yellowish grey appearance. Where islands of the normal reddish fundus background remained visible, they could resemble haemorrhages (figure 1D). RPD also faded in the vicinity of CNV (figure 1H) but they often remained visible more peripherally (figure 2A) or on the nasal side of the disc. Fading of RPD was not accompanied by granularity and calcification of their contents or focal overlying pigment changes as are associated with regression of typical drusen.

Development and spread of RPD. (A) Right eye of 58-year-old man showing moderate number of intermediate and larger drusen. (B) Age 64. Confluence of larger deposits below fixation with focal hyperpigmentation. (C) Age 70. Larger drusen have regressed to leave focal depigmentation (vertical arrow). RPD recognised as a yellowish interlacing pattern above fovea (arrowhead), separating reddish islands of normal fundus. (D) Higher magnification of another eye showing that fundus background seen through the yellowish eyeground can resemble haemorrhages. (E) Left eye of 69-year-old woman with incidental chorioretinal folds (arrowhead). RPD are not evident. (F) Age 75. RPD have developed above the fovea (arrow). The fovea itself appears spared. (G) Age 79. Red-free photograph showing that RPD have reached the superior temporal vessels (arrow). Closer to the fovea the RPD have fused (asterisk). (H) Age 82. Development of CNV. RPD no longer recognisable.

Imaging of RPD. (A) Left eye of 74-year-old woman. Retina lateral to the fovea presents a diffuse yellow appearance (arrow). Beyond this area the diffuse yellowing breaks up into RPD. (B) The yellow area is mildly hyperfluorescent on FA compared with brightly fluorescent typical drusen. (C,D) Red-free (left) and autofluorescent (right) images of right eye of woman at age 57 showing RPD and typical drusen. (E,F) At age 60 all drusen have increased in number. RPD show reduced autofluorescence separated by mildly increased autofluorescence and lie mainly in upper fundus where they extend close to disc edge. Typical drusen lie mainly below and laterally and are not well seen on autofluorescence.
RPD exhibited differences from typical drusen with various imaging modalities. RPD were more apparent in red-free and blue light, especially in fair fundi, while on FA the RPD and the diffuse yellow background that resulted from their fusion were mildly hyperfluorescent (figure 2B). Autofluorescence proved the most reliable method of distinguishing RPD (figure 2D,F), demonstrating a corresponding reticular pattern of hypoautofluorescence as described in the classification of autofluorescence by Bindewald et al5 On OCT the inner segment/outer segment junction was raised into an undulating layer but focal elevation of the RPE occurred only over typical drusen (figure 3D).

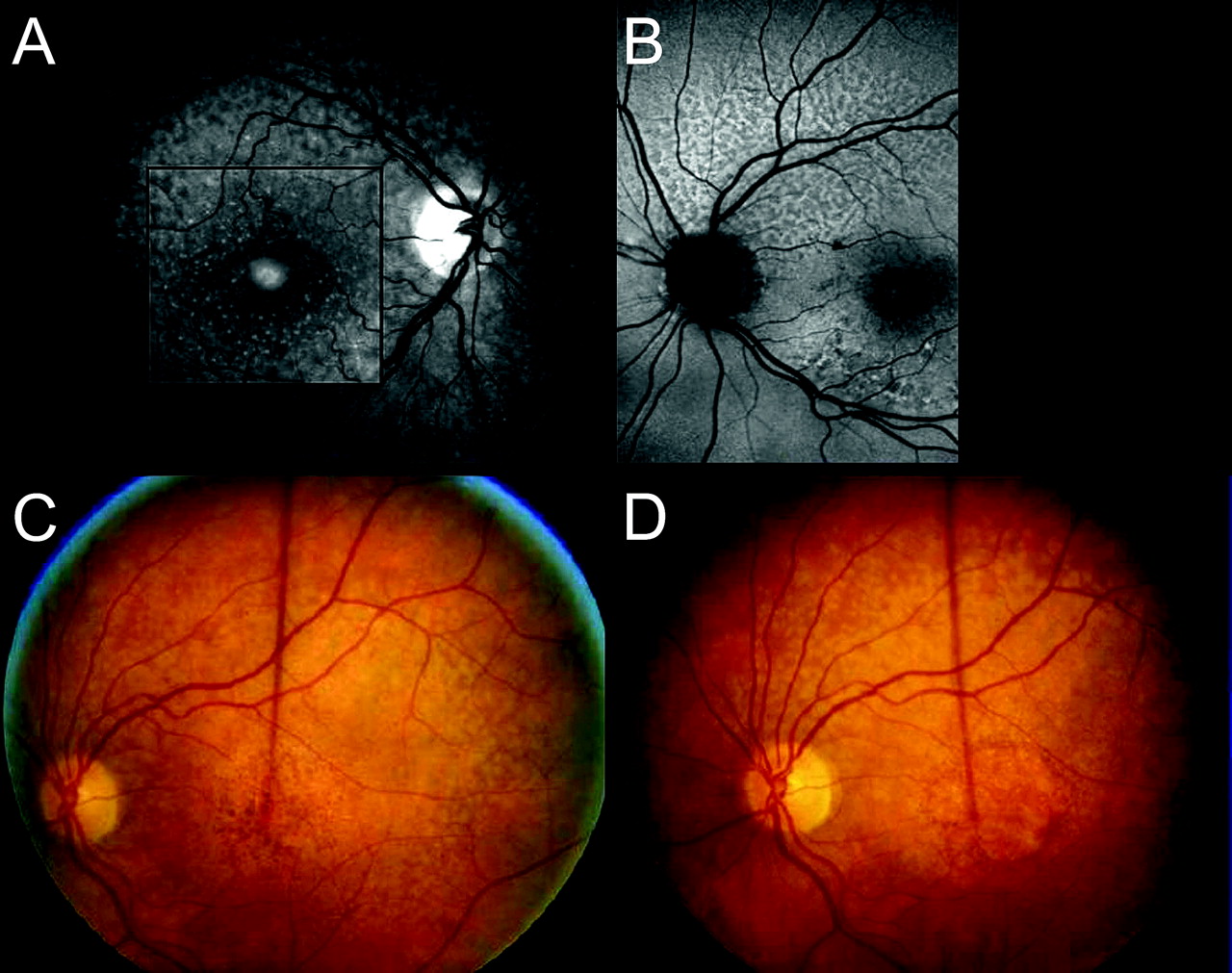
Development of RPD following onset of late AMD. (A) Right eye of 68-year-old woman with large drusen. The central drusen are fading and the RPE is attenuated (incipient atrophy). No sign of RPD. (B) Age 77. GA has developed centrally. Still no evidence of RPD which were first noted at age 79. (C) Age 82. RPD now evident superiorly. Green line indicates OCT slice which passes through two landmarks—the superior temporal vein and a large druse (d). 1=area above vein where RPD are absent. 2=area between vein and GA where RPD are prominent. 3=area of GA. (D) OCT slice shown above. In area 1 above vein (v) the RPE and photoreceptors are regular. In the area of RPD (2), arrows point to the inner segment–outer segment junction which has an undulating surface. d=druse. 3=GA.
A range of AMD associations was noted in the 332 eyes. The eyes were grouped according to the most severe manifestation of AMD present at the time that RPD were first noted (table 2). The less affected eye was designated as the study eye, RPD always being present in this eye. Only nine eyes (2.7%) had fewer than 10 small drusen and were considered normal. More than 10 discrete small or intermediate drusen were present in 57 eyes and were a pointer to the possible presence of RPD (figure 4A), similar small drusen also being noted along the distribution of RPD. An additional 122 eyes also had larger or regressing drusen and 29 eyes had hyperpigmentation around the edge of the fovea (figure 4B). Adult vitelliform lesions were present in 14 eyes (figure 4A), but were excluded as they are more commonly regarded as a dystrophy. Eyes with any evidence of CNV or with GA involving the fovea were considered as late AMD, CNV being present in 95 eyes and GA in 17 eyes, such that late AMD affected 112 eyes or approximately one-third of the total.
166 patients with reticular pseudodrusen in one or both eyes manifestations of AMD at entry into study
Associations of RPD. (A) Red-free image of right eye of 81-year-old woman with a central vitelliform lesion. RPD are prominent above. Note the small and intermediate drusen around the fovea, some also being located over the RPD. (B) Autofluorescent image of left eye of 82-year-old woman showing RPD in upper fundus extending to the disc edge. A crescent of hyperpigmentation lies around the lower margin of the fovea. (C) Left eye of woman aged 71, first seen at age 64 with a few shallow soft drusen, showing central hyper- and hypopigmentation within a diffuse yellow background. RPD can be seen around the edge. (D) Age 73. Central area of GA has developed, RPE are widespread. Other eye evolved similarly.
In the 31 eyes in which RPD developed during follow-up a simultaneous progression of AMD was often noted, including an increase in typical drusen numbers and size or evidence of drusen regression in 13 eyes (figure 1C) and development of atrophy in nine eyes. Uncommonly GA was noted before RPD were seen (figure 3). Initially GA was most often drusen related but then spread into the diffuse yellow background, at times quite rapidly (figure 4C,D).
At the conclusion of the follow-up period GA had occurred in 41 eyes. CNV was more than 4× as common as GA in this series, developing in 78 eyes during follow-up and affecting a total of 173 eyes (52.1%). At the end of the study the number of eyes with late AMD had therefore risen to 214 (64.5%), increasing from approximately one-third to two-thirds of the total.
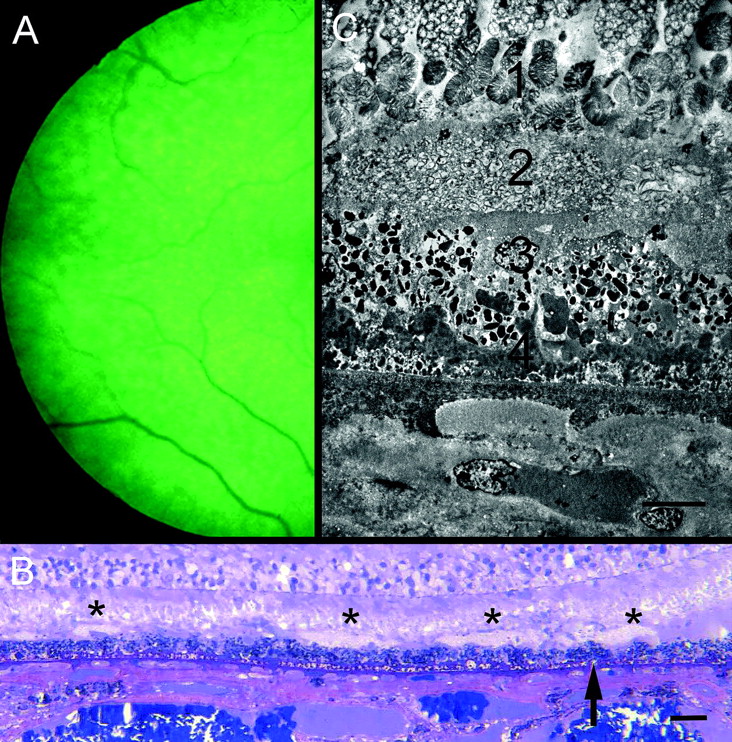
In the eye submitted to pathological examination (figure 5), RPD were most evident in the upper temporal fundus. Sections of the temporal retina demonstrated scattered collections of closely packed membrane-bound profiles on the internal surface of the RPE. Individually they measured up to 120 μm in width but this doubled where they fused. The overlying outer segments were coiled and their ends became progressively disorganised, but some outer segment profiles could be traced into these collections. The RPE was markedly abnormal with occasional proliferation into the subretinal space. External to the RPE there was a thick continuous layer of amorphous basal laminar deposit but minimal membranous debris (basal linear deposit) and only a small number of typical drusen. There was patchy loss of choroidal capillaries and the choroid was thinned but was otherwise unremarkable.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Temporal retina of right eye of 80-year-old man with early cataract. Reticular pattern evident in red-free light. Vision was 6/18. Patient died 1 year later. (B) Semithin section of temporal retina of eye shown in figure 1. Mounds of membranous debris lie on internal surface of RPE (asterisks). Basal deposits lie beneath RPE (arrow). Methylene blue-basic fuchsin stain. The scale bar indicates 40 μm. (C) Electron micrograph showing (1) undigested disorganised photoreceptor outer segments terminating in an amorphous layer; (2) subretinal deposit of membrane-bound profiles. Some outer segment profiles could be traced into these deposits; (3) irregular RPE lacking apical villi; (4) basal laminar deposit. Note loss of choroidal capillary. The scale bar indicates 10 μm.
Discussion
The full clinical significance of RPD has not been confirmed but there is increasing evidence of a high association with AMD. In the present study only 2.7% of eyes with RPD were considered otherwise normal and other studies have confirmed that there are very few eyes with RPD that have no evidence of AMD.3 4 6
The present study and that by Klein7 noted a high association with larger drusen and a high risk of late AMD. Cohen et al6 also found RPD in 24% of eyes with newly diagnosed CNV but in the CNV Prevention Trial8 RPD were seen in only 3% of fellow eyes and so were not considered a risk factor. Since an assessment of the risk posed by RPD depends upon their recognition, the present study was principally intended to review some of their distinguishing features.
One characteristic of RPD is their unusual distribution and spread. Unlike soft drusen, which have a predilection for the foveal region, RPD are most often first noted in the upper part of the macula sparing the fovea. However, at times they are also seen faintly at the fovea on autofluorescence and in near infrared light.3 As the lesions spread outwards the gaps in the network become more closely spaced and ultimately unrecognisable, resulting in an otherwise featureless yellow or yellow-grey background. RPD also fade around CNV and hence they have been considered a transient phenomenon, but often the RPD remain identifiable outside the macula. Another pointer to their presence is the finding of scattered small or intermediate drusen close to the fovea. It is unclear if these are the same as the dots described along the course of the RPD by Zweifel et al.4
RPD differ from typical drusen in other ways. Their shape in the fundus view may be sinusoidal, ring shaped or irregular and they develop as a group to form the characteristic pattern. They also present a uniform picture, unlike the variation in height, colour and density that may be exhibited by typical drusen and they have a different lifecycle without leading to focal overlying pigment changes or regression with calcification. They may extend to the termination of the RPE at the edge of the disc, whereas typical drusen uncommonly approach so closely to the disc margin.
RPD also have different imaging characteristics, especially on autofluorescence. Unlike the moderately increased autofluorescence exhibited by foveal soft drusen and the variable degree of autofluorescence possessed by other typical drusen,9 RPD consistently demonstrate decreased autofluorescence. However, Smith et al3 noted that the imaging method by which RPD were best displayed varied, some (12%) even being identified only on autofluorescence and others (5%) only on colour. He postulated this to mean that there could be preferential involvement of different retinal layers and introduced a unifying concept of ‘reticular macular disease’. Zweifel et al,4 using point-to-point correlation between autofluorescent images and spectral domain OCT, refined the location of RPD further by demonstrating that they correspond to abnormal material lying internal to the RPE and they preferred the term ‘Subretinal Drusenoid Deposits’ first introduced by Rudolf et al10 Such a subretinal location could block reflectivity from the RPE and account for the characteristic dark appearance on autofluorescence. It might also explain the undulations of the inner segment–outer segment junctions seen on OCT and the fact that underlying focal hyperpigmentation may have a grey rather than black funduscopic appearance.
The pathological specimen in the present study failed to demonstrate any evidence that RPD is due to fibrosis in the superficial choroidal stroma as previously reported,11 but in that study the retina had been missing in the single specimen examined and subretinal deposits had not been suspected. In the present study, the specimen showed instead collections of membranous material in the subretinal space. However, before accepting this as histological proof for a subretinal location for RPD, it must be pointed out that similar although smaller accumulations of subretinal membranes (1–2 RPE cells wide) are found occasionally in AMD12 without being seen clinically as RPD. Moreover, not all of this material is necessarily photoreceptor debris. Rudolf et al10 examined three eyes with such lesions and failed to demonstrate molecules associated with photoreceptors, finding instead that the material shared some markers with conventional drusen. Similar membranous debris is the hallmark of soft drusen and basal linear deposits but this term does not reflect its origin, referring only to the ultrastructural appearance of membranes with a trilaminar appearance. Curcio et al13 therefore prefer the term lipoprotein-derived debris and, since the material shares some common markers irrespective of its location, they have proposed that the RPE may release this debris from the apical as well as the basal surface.
This study therefore could support the view that RPD represent material on the inner surface of the RPE. The reason that RPD fade following CNV may have to do with migration of the RPE towards the damaged area as part of a wound-healing response.14 Possibly as the RPE becomes progressively attenuated in the process, any material associated with it would disperse. It also remains to be determined why RPD confer increased risk, the 15-year cumulative incidence of late AMD being at least double that of soft indistinct drusen.7 Zweifel et al4 therefore questioned whether microglial cells responding to debris in the subretinal space may play a role analogous to the chronic inflammatory reaction occurring beneath Bruch's membrane. Alternatively, the increased risk may simply be an indirect consequence of the fact that RPD may coincide with progression of AMD. When assessing risk a search should always be made for RPD in both eyes.
Conclusion
This study of the lifecycle of RPD indicates that their prevalence may be underestimated due to their distribution, their sometimes transient nature and confusion with typical drusen. Distinguishing features of RPD include their persistence outside the macula, their occasional close proximity to the disc, the progression to diffuse yellowing in the fundus, the coexistence of small or intermediate foveal drusen and the absence of typical signs of drusen regression. This study lends support to other reports that RPD confer heightened risk, the prevalence of late AMD increasing from one-third of eyes at baseline to two-thirds after 4.9 years. The single pathological specimen described demonstrated debris in the subretinal space.
Acknowledgments
We wish to thank Dr Alex Hunyor for his advice with early versions of the Zeiss Cirrus software.
References
Footnotes
Competing interests None declared.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the University of New South Wales Human Research Ethics Committee number 05115.
Provenance and peer review Not commissioned; externally peer reviewed.