Article Text

Abstract
Purpose Evaluation of 1-year safety profile of intravitreal ranibizumab 0.5 mg in neovascular age-related macular degeneration (NV-AMD) within routine clinical practice.
Methods The LUMINOUS programme comprises a prospective observational study assessing ranibizumab ‘real-world’ safety and clinical effectiveness across licensed indications worldwide and an annual retrospective pooled safety analysis from completed NV-AMD ranibizumab registries. 1-year data from four European registries are available. This retrospective pooled safety analysis assessed 1-year incidence rates for safety events of particular interest (key ocular or systemic events possibly related to the injection procedure or vascular endothelial growth factor inhibition) together with treatment exposure. Patients were treated according to local protocols within the ranibizumab licence.
Results Data of 4444 patients from registries in Germany (n=3470), the Netherlands (n=243), Belgium (n=260) and Sweden (n=471) were retrospectively pooled. Between 70.4% and 84.4% of enrolled patients completed 1 year of follow-up. Most frequent overall ocular events of particular interest were retinal pigment epithelial tears (27 patients; <1%) and intraocular pressure-related events (12 patients; <0.3%). Most frequent non-ocular event of particular interest was stroke (19 patients; 0.4%); annual incidence of stroke was low across all registries (0.0–0.5%).
Conclusions Ranibizumab demonstrated favourable 1-year safety profile for NV-AMD in this routine clinical practice sample, consistent with previous reported trial data. Additional data from a larger patient population are needed to better describe the long-term safety profile of ranibizumab in routine clinical practice and further evaluate risk for infrequent but serious events in ‘real-life’ settings. The 5-year LUMINOUS prospective observational study will address this need.
- Degeneration
- Macula
- Treatment Medical
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Ranibizumab (Lucentis, Novartis Pharma AG, Basel, Switzerland), a humanised monoclonal antibody fragment (Fab) specifically designed for ocular use, binds with high affinity to all isoforms of vascular endothelial growth factor (VEGF) A. Ranibizumab is licensed in Europe for treatment of neovascular (wet) age-related macular degeneration (NV-AMD), visual impairment due to diabetic macular oedema and visual impairment due to macular oedema secondary to branch or central retinal vein occlusion.
Robust evidence for clinical efficacy and safety of ranibizumab exists, supported by over 1.7 million patient treatment-years (Novartis, data on file) and evidence from more than 12 500 patients from multiple randomised, controlled clinical trials across indications.1–9
Large, long-term studies are required to further evaluate the risk of infrequent but important adverse events (such as key ocular or systemic adverse events possibly related to the injection procedure or VEGF inhibition).
Registries and observational studies provide valuable safety information and have the potential to fill important gaps in evidence because they are often larger, cover a broader range of patients, have longer follow-up periods than clinical trials and reflect real-world clinical practice.10 These benefits have led to registries and other types of observational studies becoming useful guides for healthcare decision makers.11
Pharmacovigilance programmes are required under EU legislation following approval of a pharmaceutical product and exist to monitor the safety of treatments postlaunch as they are used in routine clinical practice, providing additional information about the long-term risks, benefits and optimal use of a product. Risk management plans form part of these programmes and aim to: specify what is and is not known about safety at the time of authorisation; make a plan with milestones indicating how safety knowledge will be extended postauthorisation; where necessary, define the necessary measures to minimise known risks and monitor the success of these measures.
As part of the risk management plan for ranibizumab, the LUMINOUS programme has been designed to describe the long-term safety and effectiveness, and treatment patterns associated with ranibizumab 0.5 mg treatment in clinical practice for all approved indications included in the local product label, in a large patient population. The LUMINOUS programme consists of a retrospective and a prospective part (see online supplementary figure S1). The prospective part, the LUMINOUS study (ClinicalTrials.gov, NCT01318941), is a 5-year observational study with an expected enrolment of 30 000 patients from clinics across Asia, Australia, Europe, and North and South America. The retrospective part is an annual retrospective pooled analysis of safety data from completed, local NV-AMD ranibizumab registries.
Here, we describe the results from the retrospective pooled safety analysis of 1-year data from four completed registries in Europe. The rates of safety events of particular interest for ranibizumab, either because they could be related to the injection procedure or to VEGF inhibition, in over 4000 patients with NV-AMD are presented.
Methods
Registry selection
A retrospective analysis of pooled 1-year ranibizumab safety data from four completed ranibizumab NV-AMD registries in Europe was conducted. Included registries were those completed or with interim 1-year analysis at the time of this analysis (September 2011) (figure 1). Data were obtained from medical records of patients treated in Germany (N=3470), the Netherlands (N=243), Belgium (N=260) and Sweden (N=471). A complete list of study sites from each registry is provided in the online supplementary table S1.
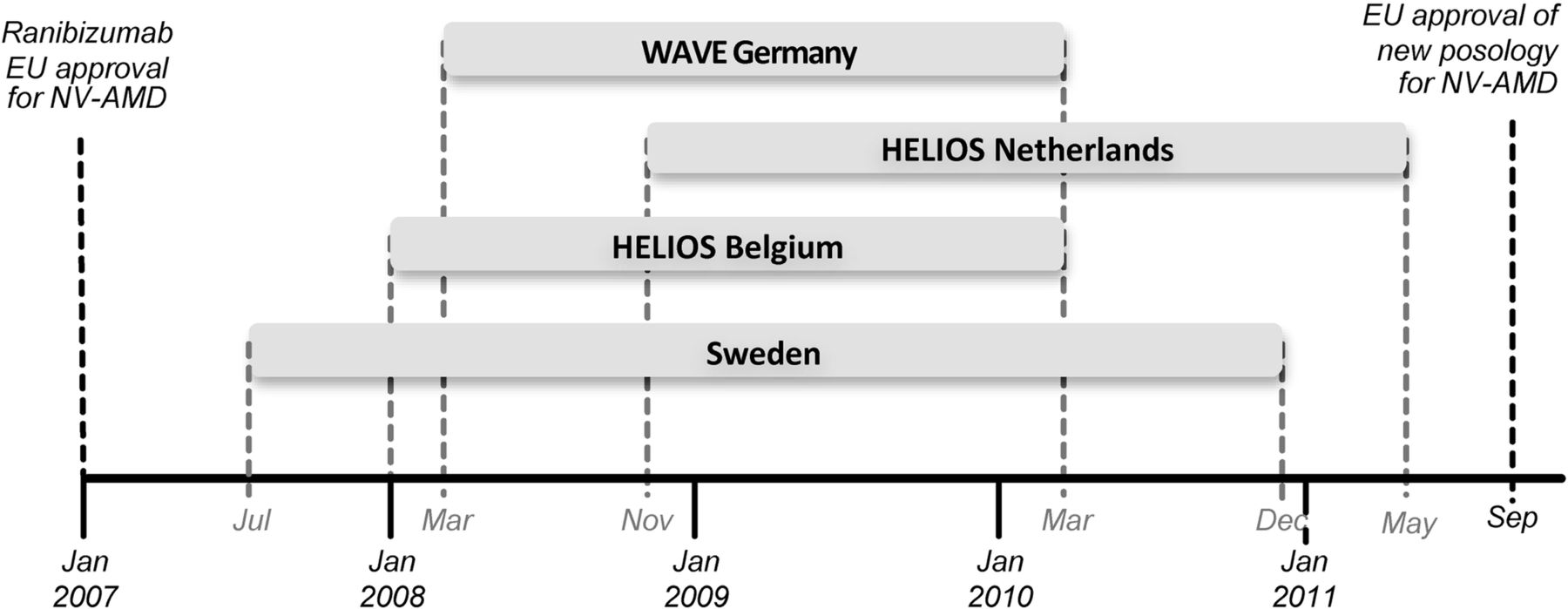
Timeline of the European registries included in the pooled analysis. Retreatment criteria were based on the European Summary of Product Characteristics during this period (>5 letter loss). The German and Swedish registries were both 1-year studies with safety among the primary objectives.13 ,14 The HELIOS studies (the Netherlands and Belgium) were 2-year studies with a primary objective to describe visual acuity outcomes over the 2-year observation period; the evaluation of the safety of ranibizumab in real life over 2 years was a secondary objective.15 16 For both HELIOS studies an interim analysis was planned once all patients had their 1-year assessment. NV-AMD, neovascular age-related macular degeneration.
Patients
Patients included in the analysis were newly diagnosed or previously diagnosed patients with recent disease progression with/without prior AMD therapy. All patients presenting during the enrolment period could be assessed for eligibility and invited to participate if they met eligibility criteria. Key inclusion criteria common to the four registries were: all adult patients with a diagnosis of NV-AMD willing and able to provide informed consent personally or by legal proxy; treatment with ranibizumab according to local practices and protocols, and within the local licensed indication of ranibizumab. The key exclusion criterion common to the four registries was concurrent participation in a clinical trial.
Patients were prospectively recruited into the registries between July 2007 and April 2010 and subsequently followed up for ≥1 year. At the time when the registries were initiated, NV-AMD was the only licensed indication for ranibizumab. Ranibizumab treatment was administered according to the European Summary of Product Characteristics, approved in 2007. Informed consent was obtained from all participants prior to study entry. Independent Ethics Committee approval was acquired as appropriate.
Data collection and reporting
In the retrospective pooled safety analysis of the four registries, 1-year incidence rates for safety events of particular interest were assessed together with treatment exposure. Data were collected at each patient visit. Timing of patient visits following baseline visit was at the discretion of the investigator in accordance with their normal patient management procedures and best clinical judgment.
The following patient demographics were recorded: patient age (at baseline); gender; visual acuity (VA) at baseline and 1 year (if available and performed as part of routine practice); cause of treatment discontinuation before the end of the 1 year observation, if applicable; number, timing and frequency of ranibizumab doses during the first year of treatment.
The reported ocular and non-ocular events were those that occurred at least once during the study period. In the pooled safety analysis, only events of particular interest were included. Events of particular interest were termed based on the mechanism of action of ranibizumab and injection procedure. Categories of safety events of particular interest and list of MedDRA (Medical Dictionary for Regulatory Activities) preferred terms for each category are shown in full in the online supplementary table S2.
Statistical analysis
Safety analyses were conducted on all patients who had received at least one dose of ranibizumab and who had at least one safety assessment following treatment. Events of particular interest were categories defined by a list of preferred terms using MedDRA V.13.0. Events from registries were internally recoded in MedDRA V.13.0 to allow allocation to the corresponding event of interest. The occurrence and proportion of patients with at least one such event of particular interest were calculated by country. Data from all countries were pooled by event of particular interest. The incidence rates for events of particular interest were derived by summating the incidence rate of the related preferred terms.
Results
Data from 4444 patients from registries in Germany, the Netherlands, Belgium and Sweden were collected and included in this retrospective safety analysis. The majority (79%) of patients in this analysis were from Germany. The percentage of enrolled patients followed up for 1 year was 74.6% in Germany (2587 of 3470), 84.4% (208 of 243) in the Netherlands, 70.4% (178 of 253 with evaluable baseline data) in Belgium and 78.6% (370 of 471) in Sweden (figure 2).

{kind=link}
{kind=link}
Patient disposition across the four registries.13–16 *Evaluable baseline (HELIOS Belgium) is the number of patients with evaluable baseline data. Safety analyses were conducted on all patients who had received at least one dose of ranibizumab and who had at least one safety assessment following treatment. N=total number of patients; n=number of patients observed. VA, visual acuity.
The mean age of patients included in the analysis was 77.6–78.7 years across the registries. The majority of patients were female. Most patients (75.1–83.5%) were treatment naïve (Germany, the Netherlands and Belgium) except for Sweden, where 57.7% patients were included retrospectively and were already receiving ranibizumab. Characteristics of patients in each registry are summarised in table 1.
Characteristics of the patients with NV-AMD
In the German, Dutch and Swedish registries, mean number of ranibizumab injections calculated for patients who completed 1 year was 4.3, 5.5 and 4.7, respectively. The mean number of injections reported in the Belgian registry was calculated for all patients and was 5.0.
During 1 year, 49 patients from HELIOS (Health Economics with Lucentis in Observational Settings) Netherlands had bilateral treatment (20.2%). In HELIOS Belgium, 13 patients had bilateral treatment at study start (5.0%). Any safety events were recorded for both eyes in these patients. Bilateral treatment data were not available for the German WAVE (Lucentis in Wet AMD: Evaluation of Visual Acuity and Quality of Life) or Swedish registries,13–16 and safety events were recorded from the study eye only.
Results for ocular and non-ocular events of particular interest are summarised in tables 2 and 3, respectively. Seventy-three ocular events and 55 non-ocular events of particular interest were recorded in 4444 patients. The most frequent ocular events were retinal pigment epithelial tear (27 patients, 0.61%) and intraocular pressure related events (12 patients, 0.27%) (table 2). Rates of retinal pigment tear and intraocular pressure related events were noticeably higher in the Netherlands than in the other countries. The most frequent non-ocular events were non-cardiac arterial thrombotic events, notably stroke (recorded as cerebral haemorrhage, cerebral infarction, cerebrovascular accident or intracranial haemorrhage; 19 patients, 0.43%) (table 3). In the Belgian registry, no strokes were reported. Across the other three registries annual incidence of stroke was similar (range 0.21–0.49%).
Summary of 1-year incidence of targeted ocular events
Summary of 1-year incidence of targeted non-ocular events
Discussion
While the efficacy and safety profile of ranibizumab in licensed indications has been demonstrated in multiple clinical trials,1–9 the safety profile of intravitreal anti-VEGF therapies in routine clinical practice remains less well established.
In a recent Medicare analysis (N=146 942), bevacizumab and ranibizumab were not associated with increased risks of mortality, myocardial infarction, bleeding or stroke compared with photodynamic therapy or pegaptanib.17 In a secondary analysis (21 815 intravitreal bevacizumab vs 19 026 ranibizumab), significantly lower risks for stroke (HR 0.78; 99% CI 0.64 to 0.96) and all-cause mortality (HR 0.86; 99% CI 0.75 to 0.98) were reported for ranibizumab compared with bevacizumab. Bevacizumab (Avastin, Roche, Basel, Switzerland) is not licensed for intraocular use or treatment of any ocular conditions.
Overall mortality with intravitreal bevacizumab compared with ranibizumab was also significantly increased in another Medicare claims analysis of >77 000 NV-AMD beneficiaries (11% greater overall mortality with bevacizumab, HR: 1.11; 99% CI 1.01 to 1.23).18 Higher risk of haemorrhagic cerebrovascular accident (HR: 1.57; 99% CI 1.04 to 2.37) was also observed for bevacizumab over 10 months, and ocular inflammation or cataract surgery after NV-AMD treatment were more likely with bevacizumab compared with ranibizumab.
In the CATT (Comparison of AMD Treatments Trial) study, serious systemic adverse events were higher with bevacizumab in year 1 compared with ranibizumab (ranibizumab 19% vs bevacizumab 24.1%, p=0.04) and persisted through year 2 (ranibizumab 31.7% vs bevacizumab 39.9%, p=0004).19 ,20 Patients receiving bevacizumab had a cumulative risk ratio of 1.30 (95% CI 1.07 to 1.57, p=0.009) for experiencing at least one systemic serious adverse event.19 For every 12 patients treated with bevacizumab instead of ranibizumab there would be one additional serious systemic adverse event (number needed to harm 12.2). The IVAN (Inhibit VEGF in Age-related choroidal Neovascularisation) trial (n=610), reported no significant differences between drugs at 1 year in serious adverse event rates (ranibizumab 9.6% vs bevacizumab 12.5%, p=0.25).21 However, the increased risk of experiencing a serious adverse event with bevacizumab observed in CATT persisted in the meta-analysis presented in the IVAN publication (p=0.016).21
Evidence that intravitreally administered anti-VEGF agents may cause sustained systemic VEGF inhibition has increased.22 ,23 VEGF-inhibitors differ in properties that may be associated with differing systemic exposure following intravitreal injection. For example, intravitreal bevacizumab has a longer systemic half-life and greater effect on plasma VEGF levels compared with ranibizumab, suggesting higher systemic drug exposure,22–24 which was also confirmed in IVAN.21
While these studies provided interesting comparative safety information of anti-VEGF therapy, limitations exist with each. To evaluate the absolute risk of infrequent, but serious side effects (such as those potentially related to systemic inhibition of VEGF) in routine clinical practice, prospective studies of large numbers of patients are required.
This pooled safety analysis of >4000 patients with NV-AMD in a routine clinical practice sample indicates that ranibizumab has a low rate of events related to the potential (systemic) effects of VEGF inhibitors or the intravitreal injection procedure. Although comparison of these observational pooled data with randomised studies must be interpreted with caution, it is interesting to draw some comparisons with other studies that adopted similar as-needed treatment dosing schedules.
The mean number of injections per patient in the analysis described here ranged from 4.3 to 5.5, similar to that observed in SAILOR (Safety Assessment of Intravitreous Lucentis fOR AMD) (4.6 in cohort 1), slightly lower than in SUSTAIN (Study of ranibizUmab in patientS wiTh subfoveal choroidal neovascularizAtIoN) (5.6) and lower than in the ranibizumab as-needed arm of the CATT study at 1 year (6.9).8 ,9 ,20 However, not all patients in the current study were treatment naïve, which could confound this comparison (75.1–83.5% treatment-naïve patients in the German, Dutch, Belgian registries and 42.3% in the Swedish registry).
The relatively low injection numbers together with suboptimal functional outcomes at 1 year suggest that patient management could be improved. The European dosing recommendation for ranibizumab in NV-AMD (since September 2011) is monthly treatment until maximum VA is achieved and treatment resumed when monthly monitoring indicates loss of VA due to NV-AMD; this should reduce the potential for undertreatment in future studies, including the prospective LUMINOUS study. A VA/optical coherence tomography (OCT)-guided pro re nata ranibizumab regimen has been shown to achieve optimal VA outcomes in patients with NV-AMD.19 ,20 ,25 Moreover, prompt initiation of treatment, plus regular monitoring for reactivation of disease and/or second eye involvement, are critical to improving outcomes. Furthermore, some incomplete responders to anti-VEGF therapy have been diagnosed with polypoidal choroidal vasculopathy following indocyanine green angiography, allowing administration of more appropriate treatment.26 ,27 Self-monitoring tools, such as the Amsler grid, are extremely useful for patients to use at home; however, more advanced and reliable tools are required.
Rates of non-ocular events that could potentially be caused by VEGF inhibition were low (all <0.5%) and in line with previously reported studies (see online supplementary table S3). The annual incidence of stroke (0.43%; including cerebral haemorrhage, cerebral infarction, cerebrovascular accident and intracranial haemorrhage) was lower than experienced with 0.5 mg ranibizumab in SAILOR (1.2%, cohort 1; including stroke, acute ischaemic stroke, intracerebral haemorrhage, cerebrovascular disease and brain haemorrhage secondary to fall) or in a 1-year analysis of 19 026 Medicare beneficiaries who received ranibizumab for NV-AMD (1.8%, secondary analysis).8 ,17 The low frequency of non-ocular events in this analysis is consistent with the low systemic exposure and short half-life of ranibizumab.28 ,29
Consistent with data from randomised clinical trials,1–3 ,20 ranibizumab was associated with a low rate of ocular events that could be caused by intravitreal injection or use of a VEGF inhibitor (see online supplementary table S3). Less than 1% of patients experienced retinal pigment epithelial tear or intraocular pressure related events and <0.3% of patients developed traumatic cataract, vitreous haemorrhage, deterioration of retinal blood flow, endophthalmitis, intraocular inflammation or retinal detachment. Annual incidence of endophthalmitis in the current analysis in clinical practice (0.11%) was lower than with 0.5 mg ranibizumab in SAILOR (0.4%; cohort 1).8
It is important to understand the complexities of registries and observational studies in order to limit potential biases. There is potential for under-reporting of safety events in observational studies. Indeed, the current analysis reported no events of proteinuria, reflecting either under-reporting or possible low systemic VEGF inhibition. The majority of patients in this study were from the German WAVE study, and underdosing of ranibizumab was common in Germany during 2007–2009. Potential undertreatment, and the relatively low number of treatments, may be one of the causes for the low number of adverse events reported in this study. To minimise any bias from under-reporting of adverse events, the prospective LUMINOUS study includes regular communications to study sites; quarterly phone calls, regular email correspondence and distribution of newsletters.
The potential for selective patient enrolment in registries can also lead to bias, towards those with greater risk for certain events or with greater disease. Participating sites in the LUMINOUS programme are advised to invite all patients who meet the inclusion criteria and consecutively enrol all patients who have given their informed consent.
The benefits of registries, including large numbers of patients across a broad population who have been treated in real-life clinical practice, ensure that the retrospective part of the LUMINOUS programme provides valuable information supplementing that already known from clinical trials about ranibizumab safety. Although most patients in this analysis were from the German WAVE registry, further national registries will be included when available. Subsequent analyses will therefore provide outcomes based on larger numbers of patients, more balanced across countries. In addition, due to lack of standardisation of data capture, eye charts used and the scope of the initial studies, retrospective data are not considered robust enough to report efficacy outcomes. However, efficacy data from the prospective arm of LUMINOUS will be available in future publications.
In conclusion, ranibizumab demonstrated a favourable safety profile in the treatment of NV-AMD over a 1-year period in real-world clinical practice. This analysis is part of the wider ongoing LUMINOUS programme, which will also collect prospective long-term safety, clinical effectiveness and health-related quality of life data on ranibizumab across all approved indications in routine clinical practice in a large, worldwide patient population.
Acknowledgments
The authors would like to thank ApotheCom, which is part of Huntsworth Health, for editorial support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Collaborators LUMINOUS Steering Committee: Prof Dr Francesco Bandello, Department of Ophthalmology, University Vita-Salute Scientific Institute, San Raffaele, Milan, Italy; Mr Christopher Brand, Royal Hallamshire Hospital, Sheffield, UK; Prof Dr Mark Gillies, Clinical Ophthalmology & Eye Health, Sydney Eye Hospital, University of Sydney, Australia; Prof Dr Frank G Holz, Department of Ophthalmology, University of Bonn, Germany; Prof Dr Adrian Koh, Eye & Retina Surgeons, Camden Medical Centre, Singapore; Prof. Dr Paul Mitchell, Department of Ophthalmology, University of Sydney, Australia; Dr Aaron Osborne, Novartis, Basel, Switzerland; Prof Dr Ursula Schmidt-Erfurth, Department of Ophthalmology, Medical University Vienna, Austria; Mr N Sharma, AMD Alliance International, Nottingham, UK; Dr Tom Sheidow, Ivey Eye Institute, Ontario, Canada; Prof Dr Eric Souied, Service d'Ophthalmologie, Hôpital Intercommunal de Creteil, Creteil, France; Dr Marta S Figueroa, Hospital Ramon y Cajal, Servicio de Oftalmologia, Madrid, Spain; Mr Steve Winyard, Royal National Institute of Blind People, London, UK.
-
Contributors All authors participated in the conception, design and implementation of the trials. All authors were involved in the interpretation of analysed data and the decision to submit for publication.
-
Funding The four registries reported in this pooled analysis were funded by Novartis.
-
Competing interests FGH is on advisory boards for Acucela, Alcon, Allergan, Bayer, GlaxoSmithKline, Genentech, Heidelberg Engineering, Optos, Novartis, and Pfizer and has received travel, honorarium and research support from these companies. FGH has no stocks, equity, contract of employment or named position on company boards. FB is on advisory boards for, provides consultancy services and gives lectures for Allergan, Novartis, Farmila-Thea Farmaceutici, Bayer, Pfizer, Alcon, Bausch and Lomb, Genentech, Alimera Sciences, Sanofi-aventis and Thrombogenics. FB has received honorarium and travel support from Novartis. FB has no stocks, equity, contract of employment or named position on company boards. MG is on advisory boards for Allergan, Novartis, Bayer and Pfizer. MG has received research support from Allergan, Bayer and Novartis, honorarium support from Allergan, Bayer, Novartis and Pfizer, and travel support from Novartis. MG has no stocks, equity, contract of employment or named position on company boards. PM is on advisory boards for Abbott, Allergan, Bayer, Novartis and Pfizer and has received travel and honorarium support from these companies. PM has no stocks, equity, contract of employment or named position on company boards. AO is an employee of Novartis Pharma AG. TS is on advisory boards for Novartis and Alcon. TS has received research support and honorarium support from Novartis. TS has no stocks, equity, contract of employment or named position on company boards. ES is on advisory boards for Bausch and Lomb, and Laboratoires Théa, provides consultancy services for Bausch and Lomb, Novartis, Allergan and Laboratoires Théa, and gives lectures for Bausch and Lomb and Novartis. ES has received travel support from Bausch and Lomb and Bayer. ES has no stocks, equity, contract of employment or named position on company boards. MSF is on advisory boards for Alcon, Allergan, Bayer and Novartis and has received travel, honorarium and research support from these companies. MSF has no stocks, equity, contract of employment or named position on company boards.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Additional unpublished data from each of the four registries included in this analysis are held by the registry representatives L. Hjelmqvist (St Erik's Eye Hospital, Karolinska Institutet, Stockholm, Sweden), FG Holz (Department of Ophthalmology, University of Bonn, Germany), JHJ Klaver (OMC Haarlem, Haarlem, The Netherlands) and J-M Rakic (Department of Ophthalmology, University of Liège, Belgium). These additional data are available, on request, from the authors of this manuscript and Novartis Pharma AG.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/