Article Text

Abstract
Aim To examine the role of microRNA-146a (miR-146a) in the regulation of fibrosis in an in vitro model of Graves’ orbitopathy (GO).
Methods Orbital fat/connective tissues were harvested from patients with GO and non-GO for primary orbital fibroblast cultures. The effects of transforming growth factor-β (TGF-β), a potent cytokine that promotes fibrosis, on miR-146a expression were analysed in GO and non-GO orbital fibroblasts using quantitative real-time PCR. The effects of overexpressed miR-146a on TGF-β-induced fibrotic markers were examined in GO orbital fibroblasts by western blot analysis. Expression ofSma and Mad related family (Smad) 4/tumour necrosis factor receptor-associated factor 6 (TRAF6) after transfection of miR-146a mimics or inhibitors were examined.
Results TGF-β induced an increase in miR-146a expression in orbital fibroblasts from patients with GO in a time-dependent and concentration-dependent manner. miR-146a mimics further decreased the production of TGF-β-induced fibronectin, collagen Iα and α-smooth muscle actin protein. The Smad4 and TRAF6 protein levels were significantly decreased by miR-146a mimics, compared with control mimics, and significantly increased on inhibition of miR-146a production compared with a control.
Conclusions miR-146a plays a role as a negative regulator in the production of TGF-β-induced fibrotic markers. Thus, miR-146a may be involved in the regulation of fibrosis in orbital fibroblasts from patients with GO.
- experimental laboratory
Statistics from Altmetric.com
Introduction
Graves’ orbitopathy (GO) is an autoimmune inflammatory disorder of the orbit.1 Although the pathogenesis of GO remains unclear, the main pathomechanism involves three mechanisms: (1) inflammation,2 (2) adipogenesis3–5 and (3) glycosaminoglycan (GAG) accumulation. In GO, GAG accumulates in extraocular muscles and orbital connective tissues.6 These conditions can lead to fibrosis as part of the wound healing process.7 Fibrosis is the formation of excess fibrous connective tissue in an organ or tissue during reparative or reactive processes.8 9 Various factors such as infection, inflammation, trauma and allergy can cause fibrosis during reparative processes.
Recently, several studies have confirmed that microRNA (miRNA) plays a critical role in the expression of genes associated with the fibrosis of organs such as heart, kidney10 and liver. miRNAs are a family of short non-coding RNAs that regulate gene expression by inhibiting their target messenger RNA (mRNA).11 12 They can participate in a wide variety of biological processes including human diseases such as cancer,13–15 inflammatory disease,2 16–19 obesity,19 infections20 21 and oxidative stress-associated disorders.22 23 Recently, we showed that the level of microRNA-146a (miR-146a) expression was significantly higher in GO orbital adipose tissue than in that of non-GO.2 In addition, we found that miR-146a was upregulated by inflammatory stress in orbital fibroblasts and reported that miR-146a may play a role in the pathogenesis of GO.
miR-146a has been relatively well studied in the field of autoimmune inflammatory disorders.2 18 19 24 It is known to inhibit the nuclear factor κ-light-chain-enhancer of the activated B-cell (NF-κB) pathway by suppressing its target mRNAs, such as those encoding tumour necrosis factor receptor-associated factor 6 (TRAF6), interleukin-1 receptor-associated kinase and Toll-like receptor 4.25 However, few studies have explored the relationship between miR-146a and fibrosis processes.10 26 Recently, Morishita et al 10 reported that miR-146a inhibited renal fibrosis by suppressing profibrotic (Smad4-transforming growth factor-β1 (TGF-β1)) and inflammation (TRAF6-NF-κB) signalling pathways.
Based on previous reports, miR-146a may inhibit orbital fibrosis by inhibiting the TGF-β signalling pathway in orbital fibroblasts. In the present study, we explored the role of miR-146a in the regulation of fibrosis in an in vitro model of GO to determine the therapeutic potential of miR-146a in GO.
Materials and methods
To test our hypothesis, orbital fat/connective tissues were harvested from nine patients with GO and three patients with non-GO and primary orbital fibroblast cultures established. We selected patients with GO who underwent surgical orbital decompression to correct for severe proptosis. Patients who had been treated with steroids and/or radiation for at least 3 months before surgery were excluded. All patients with GO were in an euthyroid state at the time of surgery. Patients with non-GO included those who underwent lower lid blepharoplasty without any prior medical history. Primary cultures of orbital fibroblasts were performed according to the methods described previously.2 7 27 Written informed consent was obtained from all subjects.
To explore the effect of TGF-β (Cell Signaling Technology, Danvers, Massachusetts, USA) (a potent cytokine-promoting fibrosis)8 9 on miR-146a expression, orbital fibroblasts from GO and non-GO tissues were plated onto six-well plates for measurement of RNA levels.
Cells were stimulated in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 (DMEM/F-12) with the indicated TGF-β concentration (0, 1, 2.5, 5 and 10 ng/mL) or with 5 ng/mL TGF-β for the indicated time (0, 3, 6, 16 and 24 hours). miR-146a expression under different conditions was investigated using quantitative real-time PCR (qRT-PCR). We compared the effects of TGF-β on miR-146a expression in GO and non-GO orbital fibroblasts.
Additionally, we explored the effect of TGF-β on the phosphorylation of several cytoplasmic mediators; orbital fibroblasts from GO were stimulated in DMEM/F-12 with 5 ng/mL TGF-β for the indicated times (0, 5, 15, 30, 60 and 180 min). The expression of cytoplasmic mediators was investigated using western blot analysis. The following mediators were used in the present study: extracellular signal-regulated kinases (ERK), phospho-ERK, p38, phospho-p38, NF-κB, phospho-NF-κB, c-Jun N-terminal kinase (JNK), phospho-JNK, v-Akt murin thymoma viral oncogene (AKT), phospho-AKT, phosphoinositide-3 kinase (PI3K), phospho-PI3K, Sma and Mad related family (Smad) 2, phospho-Smad2, mechanistic target of rapamycin (mTOR) and phospho-mTOR. All antibodies were purchased from Cell Signaling Technology.
Furthermore, we investigated whether inhibitors of ERK (PD098059; Sigma-Aldrich, St. Louis, Missouri, USA), p38 (SB203580; Sigma-Aldrich), NF-κB (SC-514; Sigma-Aldrich), JNK-1/2 (SP600125; Sigma-Aldrich), PI3K/AKT (LY294002; Sigma-Aldrich), Smad2 (SB431542; Sigma-Aldrich) and mTOR (rapamycin; AG Scientific, San Diego, California) effectively blocked the appropriate pathways in GO orbital fibroblasts; we used western blotting to this end. Each inhibitor was added at the indicated concentration 60 min prior to the addition of TGF-β (5 ng/mL, 15 min). The inhibitor concentrations were as follows: ERK (PD: 20 µM), p38 (SB2: 20 µM), NF-κB (SC: 10 µM), JNK-1/2 (SP: 20 µM), PI3K/AKT (LY: 20 µM), Smad2 (SB4: 10 µM) and mTOR (rapamycin: 0.1 µM).
Next, to explore the molecular mechanism of TGF-β-induced miR-146a expression, the effects of inhibitors of ERK (PD: 20 µM 60 min), p38 (SB2: 20 µM 60 min), NF-κB (SC: 10 µM 60 min), JNK-1/2 (SP: 20 µM 60 min), PI3K/AKT (LY: 20 µM 60 min), Smad2 (SB4: 10 µM 60 min) and mTOR (rapamycin: 0.1 µM 60 min) were investigated using GO orbital fibroblasts. To explore effects on TGF-β-induced miR-146a expression, the inhibitors were added at the indicated concentrations 60 min prior to the addition of TGF-β (5 ng/mL, 16 hours).
Additionally, to compare between TGF-β-induced miR-146a expression and interleukin (IL)-1β-induced miR-146a expression, GO orbital fibroblast were treated with IL-1β (10 ng/mL) and the expression of miR-146a was determined at 16 hours.
Transfection of orbital fibroblasts with miR-146a mimics, inhibitors and control was performed according to the respective manufacturer’s protocol.2 The miR-146a mimics and inhibitors were purchased from Ambion/Applied Biosystems (MC10722; Ambion, Austin, Texas, USA) and Exiqon (4103092–001: Exiqon, Vedbaek, Denmark), respectively. Lipofectamine 2000 Reagent (Life Technologies, Carlsbad, California, USA) was used during transfection procedure. A negative control mimic (catalogue no 4464058) was purchased from Invitrogen. A negative control inhibitor (199 006–001, TAACACGTCTATACGCCCA) was purchased from Exiqon.
Orbital fibroblasts were plated onto six-well plates to assess cytokine release. Cells were transfected with the indicated miR-146a mimics at concentrations of 30 nM using Lipofectamine 2000 Reagent. The effects of miR-146a mimics on TGF-β-induced fibrotic markers such as fibronectin (FN; BD Bioscience, Franklin Lake, New Jersey, USA), collagen Iα (Santa Cruz Biotechnology, Santa Cruz, California) and α-smooth muscle actin (SMA; Sigma-Aldrich) production were investigated using western blotting.
Previous reports have indicated that Smad4 and TRAF6 are targets of miR-146a. However, the situation in human orbital fibroblasts has not been investigated. Therefore, we explored the relationship between Smad4/TRAF6 expression and miR-146a status in such fibroblasts. To identify the fibrotic targets of miR-146a in GO orbital fibroblasts, we used western blotting to quantify Smad4 and TRAF6 expression after transfection of miR-146a mimics/inhibitors (30 nM).
qRT-PCR and western blotting
qRT-PCR and western blotting were performed as described previously.2 Briefly, for qRT-PCR, total RNA was extracted from orbital fibroblasts according to manufacturer’s instructions. 1 µg of RNA was used to generate cDNA using a TaqMan miRNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, California, USA). The cDNA was amplified using a thermocycler (ABI StepOnePlus Real-Time PCR; Applied Biosystems) with TaqMan universal PCR master mix (No AmpErase UNG; Applied Biosystems). RNU6B expression was used for normalisation. All PCRs were performed in triplicate. The results are presented as relative fold changes of threshold cycle (Ct) relative to the control group, determined using the 2−ΔΔCt method.28
Western blotting was performed as follows: Orbital fibroblasts were washed with ice-cold phosphate-buffered saline. For tissue preparation, cell lysis buffer (20 mM HEPES, pH 7.2, 10% (vol/vol) glycerol, 10 mM Na3VO4, 50 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 0.1 mM dithiothreitol, 1 µg/mL leupeptin, 1 µg/mL pepstatin and 1% (vol/vol) Triton X-100) was used. Lysates were harvested by 30 min of incubation in cell lysis buffer. Reagents were purchased from Sigma-Aldrich. Lysates were centrifuged for 10 min at 12 000×g, after which the cell homogenate fractions were stored at −70°C until use.
Equal amounts of protein (50 µg) were boiled in sample buffer. Next, 10% (wt/vol) sodium dodecyl sulfate polyacrylamide gel electrophoresis was used for the separation of each protein. Proteins were transferred onto polyvinylidene difluoride membranes (Immobilon; Millipore, Billerica, Massachusetts, USA). The samples were probed overnight with primary antibodies in Tris-buffered saline containing Tween 20 (TBST) and washed three times with TBST. Immunoreactive bands were detected with horseradish peroxidase-conjugated secondary antibody and developed using an enhanced Chemiluminescence Kit (Amersham Pharmacia Biotech, Piscataway, New Jersey, USA) and finally exposed to X-ray film (Amersham Pharmacia Biotech). Immunoreactive bands were quantified based on densitometry and normalised relative to the β-actin level of the same sample.
Statistical analysis
All experiments were performed in duplicate on at least three orbital fibroblast lines isolated from various patients with GO or non-GO samples. Means and SDs were calculated after normalisation of the miRNA and protein levels measured in at least three samples harvested from different individuals. Differences between groups were assessed by both the independent and paired t-test. A P value <0.05 was considered to reflect statistical significance.
Results
Effects of TGF-β on miR-146a expression in GO and non-GO orbital fibroblast
We measured the effects of TGF-β on miR-146a expression in GO and non-GO orbital fibroblasts exposed to TGF-β. miR-146a expression increased in a time-dependent and concentration-dependent manner on treatment of GO but not non-GO orbital fibroblasts (figure 1).

Effects of transforming growth factor-β (TGF-β) on microRNA-146a (miR-146a) expression in Graves’ orbitopathy (GO) and non-GO orbital fibroblasts. (A) concentration-dependent change of miR-146a, (B) time-dependent change of miR-146a. miR-146a expression increased in a time-dependent and concentration-dependent manner on treatment with TGF-β (*P<0.05, vs time-matched and concentration-matched controls) in GO orbital fibroblasts. Non-GO cells showed no response on miR-146a expression following TGF-β stimulation, different from GO cells. The results are expressed as mean±SD of at least three individual samples and the graphs are representative of three independent experiments. h, hours.
Effects of TGF-β on the phosphorylation of cytoplasmic mediators in GO orbital fibroblast
The phosphorylation of proteins involved in TGF-β signalling in orbital fibroblasts from patients with GO was measured after exposure to TGF-β. TGF-β (5 ng/mL) increased the phosphorylation of ERK, p38 and JNK with a peak at 30 min after pretreatment. Phosphorylation of NF-κB, PI3K, Smad2 and mTOR increased with a peak at 60 min after pretreatment. The phosphorylation of AKT showed an increase after 160 min of treatment. TGF-β resulted in an increase in the phosphorylation of proteins involved in TGF-β signalling in a time-dependent manner (figure 2).

Effects of transforming growth factor-β (TGF-β) on phosphorylation of cytoplasmic mediators in Graves’ orbitopathy (GO) orbital fibroblast. Representative blots (left column) and quantification by densitometry, normalised to the β-actin level are shown for each cytoplasmic mediators in GO orbital fibroblasts (right column). The data in the column are the mean relative density/β-actin fold ±SD (black: phospho-cytoplasmic mediators, grey: cytoplasmic mediators, respectively). ERK, extracellular signal-regulated kinases; JNK, c-Jun N-terminal kinase; mTOR, mechanistic target of rapamycin; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cell; PI3K, phosphoinositide-3 kinase; Smad, Sma and Mad related family; AKT, v-Akt murin thymoma viral oncogene.
Effects of inhibitors of TGF-β signalling on the phosphorylation of cytoplasmic mediators in GO orbital fibroblast
We used western blotting to investigate whether inhibitors of ERK, p38, NF-κB, JNK-1/2, PI3K/AKT, Smad2 and mTOR effectively blocked the appropriate pathways in GO orbital fibroblasts. TGF-β increased the phosphorylation of all of ERK, p38, NF-κB, JNK-1/2, PI3K/AKT, Smad2 and mTOR, and this was inhibited by the relevant inhibitors (figure 3).

Effects of inhibitors of transforming growth factor-β (TGF-β) signalling on the phosphorylation of cytoplasmic mediators in Graves’ orbitopathy orbital fibroblasts. Representative blots (left column) and densitometric quantifications (normalised to the β-actin levels) for each cytoplasmic mediator (right column). The figures in the latter column are the mean fold relative densities compared with those of β-actin, ±SDs (black: phosphorylated cytoplasmic mediators, grey: non-phosphorylated mediators). ERK, extracellular signal-regulated kinases; JNK, c-Jun N-terminal kinase; LY, phosphoinositide-3 kinase inhibitor; mTOR, mechanistic target of rapamycin; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PD, ERK inhibitor; PI3K, phosphoinositide-3 kinase; RA, mechanistic target of rapamycin inhibitor; SB2, p38 MAP kinase inhibitor; SB4, Smad2 inhibitor; SC, nuclear factor κ-light-chain-enhancer of activated B-cell inhibitor; Smad, Sma and Mad related family; SP, c-Jun N-terminal kinase-1/2 inhibitor.
Effects of inhibitors of TGF-β signalling on TGF-β-induced miR-146a expression in GO orbital fibroblast
For further study of the molecular mechanism of TGF-β-induced miR-146a expression, the effects of inhibitors of ERK, p38, NF-κB, JNK-1/2, PI3K/AKT, Smad2 and mTOR were measured in orbital fibroblasts of patients with GO. All inhibitors of TGF-β signalling significantly inhibited this increase in miR-146a (all P values <0.05) (figure 4). IL-β induced more miR-146a expression than did TGF-β (figure 4).

Effects of inhibitors of transforming growth factor-β (TGF-β) signalling on TGF-β-induced microRNA-146a (miR-146a) expression in Graves’ orbitopathy orbital fibroblasts. The increase in miR-146a expression induced by TGF-β was inhibited by all inhibitors of extracellular signal-regulated kinases, p38, nuclear factor κ-light-chain-enhancer of activated B cell, c-Jun N-terminal kinase-1/2, phosphoinositide-3 kinase/AKT, Smad2 and mechanistic target of rapamycin phosphorylation (*P<0.05, vs miR-146a expression after TGF-β exposure). The results are expressed as mean±SD of three individual samples and the graphs are representative of three independent experiments. LY, phosphoinositide-3 kinase inhibitor; PD, ERK inhibitor; RA, mechanistic target of rapamycin inhibitor; SB2, p38 MAP kinase inhibitor; SP, c-Jun N-terminal kinase-1/2 inhibitor; SC, nuclear factor κ-light-chain-enhancer of activated B-cell inhibitor; SB4, Smad2 inhibitor.
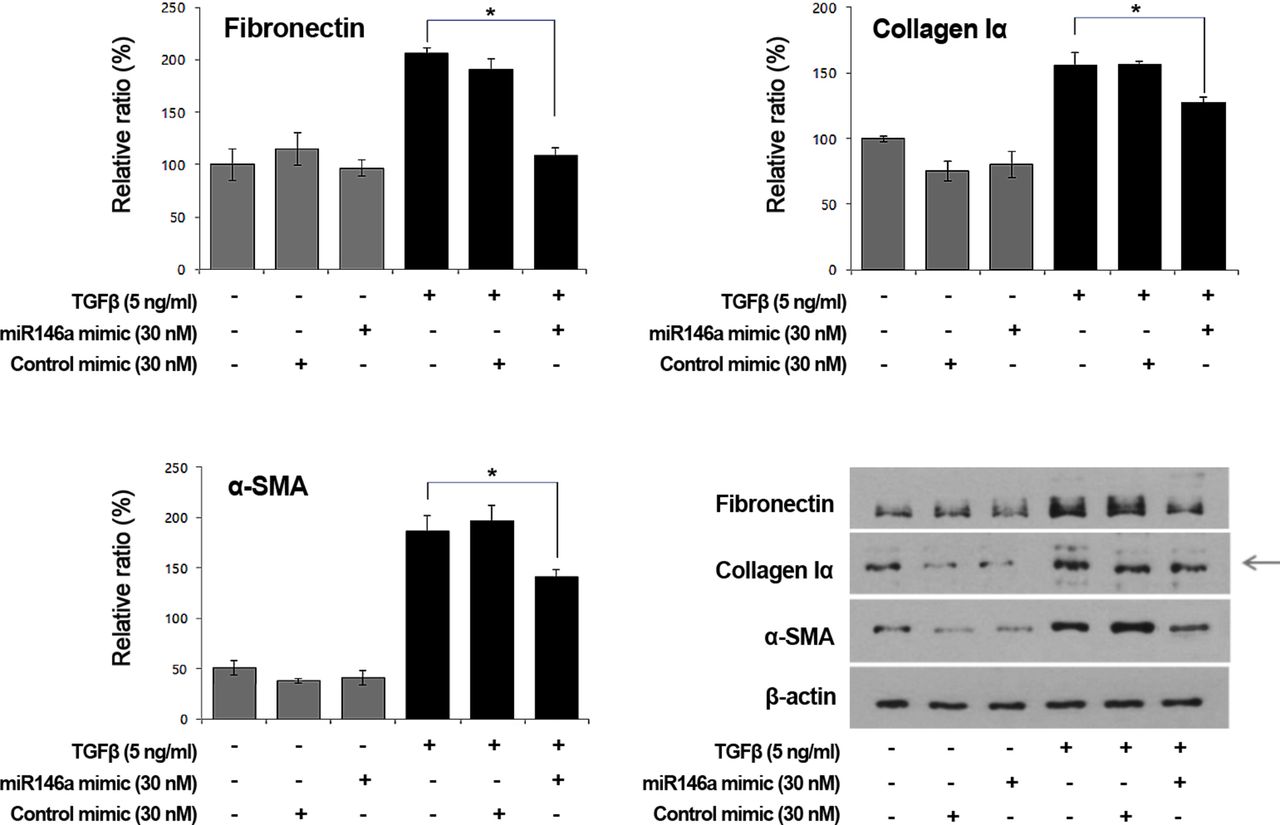
Effects of miR-146a mimics on TGF-β-induced FN, collagen Iα and α-SMA protein production in GO orbital fibroblast
We additionally examined the effects of miR-146a mimics on the expression of fibrotic markers such as FN, collagen Iα and α-SMA in orbital fibroblasts from patients with GO using western blotting. FN, collagen Iα and α-SMA expression was increased after TGF-β stimulation and increases in FN, collagen Iα and α-SMA were inhibited by miR-146a mimics (30 nM) (figure 5).

Effects of microRNA-146a (miR-146a) mimics on transforming growth factor-β (TGF-β)-induced fibronectin (FN), collagen Iα and α-smooth muscle actin (SMA) production in Graves’ orbitopathy orbital fibroblasts. FN, collagen Iα and α-SMA expression increased after TGF-β stimulation and the increases were inhibited by miR-146a mimics (30 nM) (*P<0.05 vs the FN, collagen Iα and α-SMA production levels after TGF-β exposure). The bar graphs show the means±SDs of data from at least three individual samples and the graphs are representative of three independent experiments. Representative western blotting data are shown.
Direct targets of miR-146a in GO orbital fibroblasts
The levels of both the Smad4 and TRAF6 proteins decreased significantly after transfection of miR-146a mimics, compared with control mimics and Smad4 and TRAF6 expression increased significantly on miR-146a-mediated inhibition compared with addition of control inhibitors (figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Smad4 and tumour necrosis factor receptor-associated factor 6 (TRAF6) protein expression following exposure to microRNA-146a (miR-146a) mimics or inhibitors. Graves’ orbitopathy orbital fibroblasts were transfected with either miR-146a mimics or inhibitors and the expression of Smad4 and TRAF6 protein were detected. The results of bar graph are expressed as mean±SD of at least three individual samples and the graphs are representative of three independent experiments. Representative data of western blot analysis are shown.
Discussion
In this study, we hypothesised that miR-146a inhibits orbital fibrosis by inhibiting the TGF-β signalling pathway in orbital fibroblasts. As shown by our results, miR-146a expression was increased by TGF-β in a time-dependent and concentration-dependent manner in orbital fibroblasts harvested from patients with GO. We also found that the TGF-β-induced increase in miR-146a expression was inhibited by inhibiting ERK, p38, JNK, NF-κB, mTOR and Smad2 phosphorylation. Furthermore, TGF-β-induced FN, collagen Iα and α-SMA protein production was decreased by miR-146a mimics. These results indicated that miR-146a may be involved in the production of TGF-β-induced fibrotic markers as a negative regulator.
To date, many studies have explored the question of whether miR-146a expression is increased in diseased tissues.17 26 29 Recently, we showed that expression of miR-146a was significantly higher in GO than non-GO orbital adipose tissues.2 In a previous study, we focused on the role played by miR-146a in the regulation of inflammation in GO and suggested that miR-146a may positively affect the anti-inflammatory process.2 Likewise, miR-146a has been shown to be upregulated in many autoimmune diseases and plays a key role in negative feedback. However, the role played by miR-146a in GO may be a little more complex. In contrast to the results of our previous study, Wang et al 30 reported that miR-146a exerted a pro-inflammatory effect (thus increasing IL-6 production), inhibiting the miR-146a target gene notch2, leading to exacerbation of GO, which is consistent with the findings of our previous study.30 Wei et al 31 reported that the circulating miR-146a level was significantly decreased in GO compared with non-GO subjects. We believe that such contrary results require further elucidation. Although experimental data are lacking, Li et al suggested that increased miR-155 and decreased miR-146a levels may promote ocular inflammation and proliferation in those with GO. Therefore, other miRNAs may play additional roles and their proportions may be of clinical importance.32
To support the role of miR-146a as the therapeutic target, we explored the antifibrotic effect of miR-146a in an in vitro culture system of GO during this time. Few studies have explored the relationship between miR-146a and fibrosis; Sonkoly et al 26 reported that miR-146a was significantly overexpressed in psoriatic skin lesions, which is a well-known fibrosing skin disease, compared with normal skin. Morishita et al 10 reported that miR-146a inhibited renal fibrosis by suppressing the profibrotic (Smad4-TGF-β1) and inflammation (TRAF6-NF-κB) signalling pathways. In the present study, we found that miR-146a mimics significantly decreased TGF-β-induced FN, collagen Iα and α-SMA protein production.
Activation of TGF-β signalling leads to the activation of ERK of the non-Smad pathway and Smad2/4 of the Smad pathway.9 In the present study, the phosphorylation of proteins involved in TGF-β signalling was measured following TGF-β exposure in orbital fibroblasts from patients with GO. As expected, TGF-β caused increases in the phosphorylation of ERK, p38, JNK, NF-κB, PI3K, AKT, Smad2 and mTOR. TGF-β time-dependently increased the phosphorylation of TGF-β signalling proteins in orbital fibroblasts. Furthermore, we explored the molecular mechanism underlying TGF-β-induced miR-146a expression. The effects of inhibitors of ERK, p38, NF-κB, JNK-1/2, PI3K/AKT, Smad2 and mTOR were analysed using GO orbital fibroblasts. TGF-β-induced increases in miR-146a expression were significantly inhibited by all inhibitors of TGF-β signalling (all P values <0.05). These results indicated that TGF-β-induced increases in miR-146a expression were inhibited through the ERK and Smad pathways, suggesting that the activation of ERK and Smad pathways is required for TGF-β-induced miR-146a expression in orbital fibroblasts.
Similar to miR-146a, Tan et al 33 reported that miR-29 mediates TGF-β-induced extracellular matrix synthesis in human orbital fibroblasts. Contrary to miR-146a, miR-21 is known to promote fibrosis in orbital fibroblasts in thyroid-associated ophthalmopathy. Likewise, more than 3000 mature miRNAs are the key regulators of many biological processes.19 33 They control protein expression by inhibiting the translation and degradation of target mRNAs. According to previous studies, Smad4 may be a target gene of miR-146a. Morishita et al 10 showed that the exogenous delivery of miR-146a inhibits the TGF-β/Smad and TRAF6-NF-κB signalling pathway, resulting in attenuation of renal fibrosis. The authors suggested that miR-146a may be a therapeutic option for end-stage renal disease. Sun et al 34 have recently reported that miR-146a acts as a negative regulator of TGF-β signalling in skeletal muscle after acute contusion. In the present study, we confirmed that Smad4 and TRAF6 might also be target genes of miR-146a in orbital fibroblasts.
In conclusion, miR-146a plays a role as a negative regulator in the production of TGF-β-induced fibrotic markers. miR-146a may be involved in the regulation of fibrosis in orbital fibroblasts from patients with GO. However, additional studies are required to further examine the potential of miR-146a as a therapeutic target.
References
Footnotes
Contributors SYJ and JSY were responsible for the study conception and design, as well as the intellectual content of the paper. SJP made intellectual contributions to the text. MKC performed experiments. JHL and EJL revised the article critically for intellectual content. All authors read and approved the final manuscript.
Funding This study was supported by a faculty research grant from Yonsei University, College of Medicine (grant no. 6-2015-0046 to JSY) and by the National Research Foundation of Korea (NRF-2017R1A1A1A05001051).
Competing interests None declared.
Patient consent Obtained.
Ethics approval This study was approved by the Institutional Review Board of the Severance Hospital, Yonsei University College of Medicine.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- At a glance