Article Text

Abstract
AIM To confirm the mutation of the keratoepithelin gene in patients with a severe form of superficial juvenile granular corneal dystrophy (GCD).
METHODS Five Japanese probands in whom GCD was diagnosed after histopathological examination and who developed severe manifestations of GCD in their first decade of life were investigated. Other affected family members of two probands were also examined. All probands were the offspring of consanguineous parents. DNA was extracted from their peripheral blood leucocytes and mutational analysis of the gene was performed by the polymerase chain reaction and direct sequencing.
RESULTS Four of the five probands underwent their first keratectomy or keratoplasty in their teens and subsequently underwent a second or third keratoplasty. Each proband had a homozygous G → A transition at codon 124, replacing Arg → His, of the keratoepithelin gene. Their moderately affected family members were heterozygous for the mutation.
CONCLUSIONS This finding suggests that the severity of the corneal phenotype depends on the dose effect of the mutant gene.
- Avellino corneal dystrophy
- keratoepithelin
- R124H mutation
- homozygote
Statistics from Altmetric.com
Granular corneal dystrophy (GCD) is inherited in an autosomal dominant manner and becomes manifest in the first decade of life.1 This condition is characterised by corneal granular opacities that gradually enlarge, increase in number, aggregate, and spread into the deeper and mid peripheral stroma. Most individuals with GCD maintain their vision for many years, and require keratoplasty only in the sixth or seventh decade. However, a small percentage of patients experience rapid progression of clinical manifestation and visual deterioration with an early onset around 6 years of age, and require keratoplasty in the first or second decade.2-9 Such atypical and more severe form of GCD have been described as a variant of superficial GCD. Two types of severe corneal conditions are found at an early age. One type shows fine diffuse subepithelial opacification, with a higher incidence of painful corneal erosions.3 4No consanguineous marriage was described in their families. The other type shows confluent dense white opaque corneal opacities, with painful corneal erosions being rare.2 5-9 The latter young patients are probably homozygous for the dystrophy gene as a result of consanguineous parentage.2 5 6 9
Recent molecular genetic analysis has identified missense mutations at codon 124 (Arg → His) and codon 555 (Arg → Trp) of the human transforming growth factor β induced gene (BIGH3) in individuals with granular lattice corneal dystrophy (Avellino corneal dystrophy, or ACD) and GCD, respectively.10 These monogenic disorders manifest clinically in the presence of a mutant keratoepithelin, the product of BIGH3, in the heterozygous state. We have previously reported that, in Japan, GCD accompanied by amyloid deposits (that is, ACD) and associated with the R124H mutation is most common,11 whereas GCD associated with the R555W mutation is rare. Clinically, ACD has been diagnosed incorrectly as GCD in Japan,12 because of the presence of sharply demarcated, greyish white opacities in the anterior central stroma, red staining observed with Masson’s trichrome on histological evaluation, or the presence of rod-shaped structures observed on electron microscopic examination.1
In the present study, we have confirmed that ACD, in five unrelated Japanese patients with the severe form of ACD with an early onset, was caused by homozygous R124H mutation of keratoepithelin. All five patients were the offspring of consanguineous parents.
Patients and methods
CASE REPORTS
Case 1
The proband (IV-10 in Fig 1, bottom right), a 20 year old Japanese woman, was first seen by her ophthalmologist at the age of 2 years because her parents noticed whitish corneal opacities in both her eyes. In 1987, at 10 years of age, she was referred to Keio University Hospital for severe corneal opacities that substantially impaired her vision in both eyes.13 Slit lamp examination revealed bilateral dense nodular epithelial and anterior stromal opacities, which left only a peripheral rim of clear cornea (Fig 1, top left). The area between lesions was clear, but the lesions were too numerous and close together to count. The best corrected vision in each eye was 20/200. She had no corneal erosive episodes. The corneal appearances of the other five affected individuals in her family (her brother, parents, and grandmothers) in 1987 were previously reported elsewhere.13 She was diagnosed as having a severe variant of juvenile GCD, and as probably homozygous for the responsible mutation because both of her parents were first cousins and had GCD (Fig 1, bottom right). She underwent a lamellar keratoplasty in both eyes in 1987. One year after surgery, whitish opacities were apparent in the subepithelial region. Nine years after surgery, visual acuity was 20/60 in the right eye and 20/90 in the left eye due to severe recurrence of the disease (Fig 1, bottom left). She again underwent lamellar keratoplasty in the left eye in March 1996, and in the right eye in March 1997. Slit lamp photographs of the affected corneas of her brother and parents in 1997 are shown in Figure 1. Her brother (IV-9) and father (III-6) showed only several discrete grey white opacities (Fig 1, top right and second row left, respectively). Her mother (III-5) showed several discrete grey white opacities typical of GCD in the subepithelial stroma and a few whitish fusiform or stellate opacities (Fig 1, second row right). Unlike case 1, corneal appearances of these three individuals progressed slightly during 10 years.

Slit lamp photographs of the corneas of affected family members in case 1. Top left, the proband at age 10 years before surgery (IV-10). Top right, the proband’s brother at 21 years of age (IV-9). Second row left, the proband’s father at 46 years (III-6). Second row right, the proband’s mother at 48 years (III-5). Bottom left, the proband 9 year after the first surgery. Bottom right, Pedigree of the family of case 1. The genotype with regard to the R124H mutation in the BIGH3 gene is indicated: +/+, homozygous mutation; +/−, heterozygous mutation; −/−, wild type.
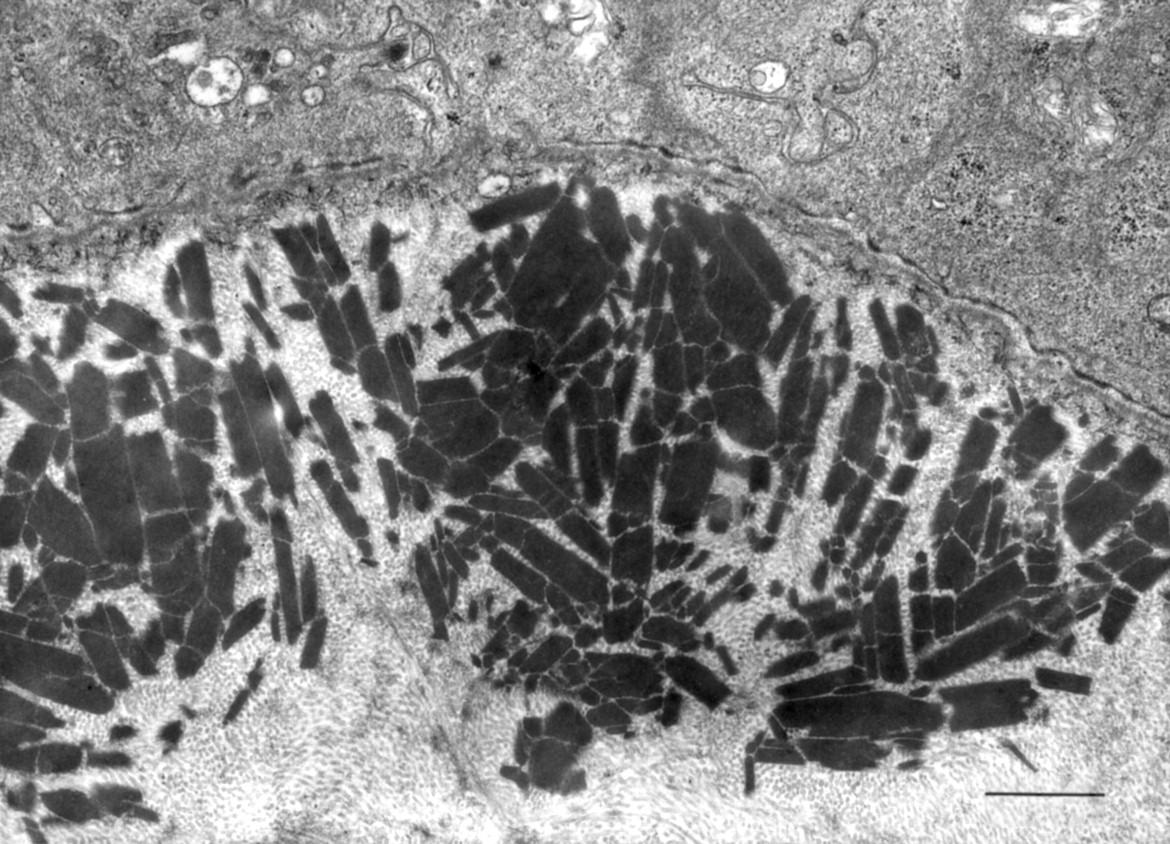
Histological analysis of corneal specimens obtained from the proband in 1987 and 1997 revealed aggregates of eosinophilic deposits that occupied the anterior-most portion of the stroma. Paraffin sections stained with Masson’s trichrome showed these deposits to have a red granular appearance. The deposits also stained moderately with Congo red, but, in the specimens obtained at 10 and 20 years of age, they did not exhibit birefringence and dichroism under polarised light. Electron microscopy revealed multiple electron dense, rod-shaped granules characteristic of GCD present in the superficial stroma adjacent to the epithelial basement membrane (Fig 2).

Electron microscopy of the cornea of case 1. Subepithelial and superficial stromal granules. Scale bar, 1 μm.
Case 2
The parents of this 47 year old Japanese woman first noticed whitish corneal opacities in both of her eyes when she was 5 years old. These opacities gradually increased in size and number, and by the age of 12 years, the patient’s visual acuity was reduced to 20/200 in each eye. She was diagnosed as having GCD since both parents also showed granular corneal opacities. She was the offspring of consanguineous parents—that is, an uncle and his niece. She was referred to Keio University Hospital for the treatment of her corneal disorder at the age of 13 years. Slit lamp examination then revealed bilateral dense nodular epithelial and anterior stromal opacities, which were too numerous and close together to count, and that left only a peripheral rim of clear cornea (Fig 3, top). She underwent lamellar keratoplasty in the left eye at the age of 13 years and in the right eye at the age of 14 years. The patient discontinued coming to the hospital until she was 27 years old, when the visual acuity in both eyes had decreased to finger counting as a result of severe recurrence after grafting (Fig 3, bottom). She underwent a penetrating keratoplasty in each eye at the age of 27 and 28 years, respectively. At the age of 47 years, her visual acuity had decreased to 20/200 in both eyes because of recurrence after grafting. She had no corneal erosive episodes.
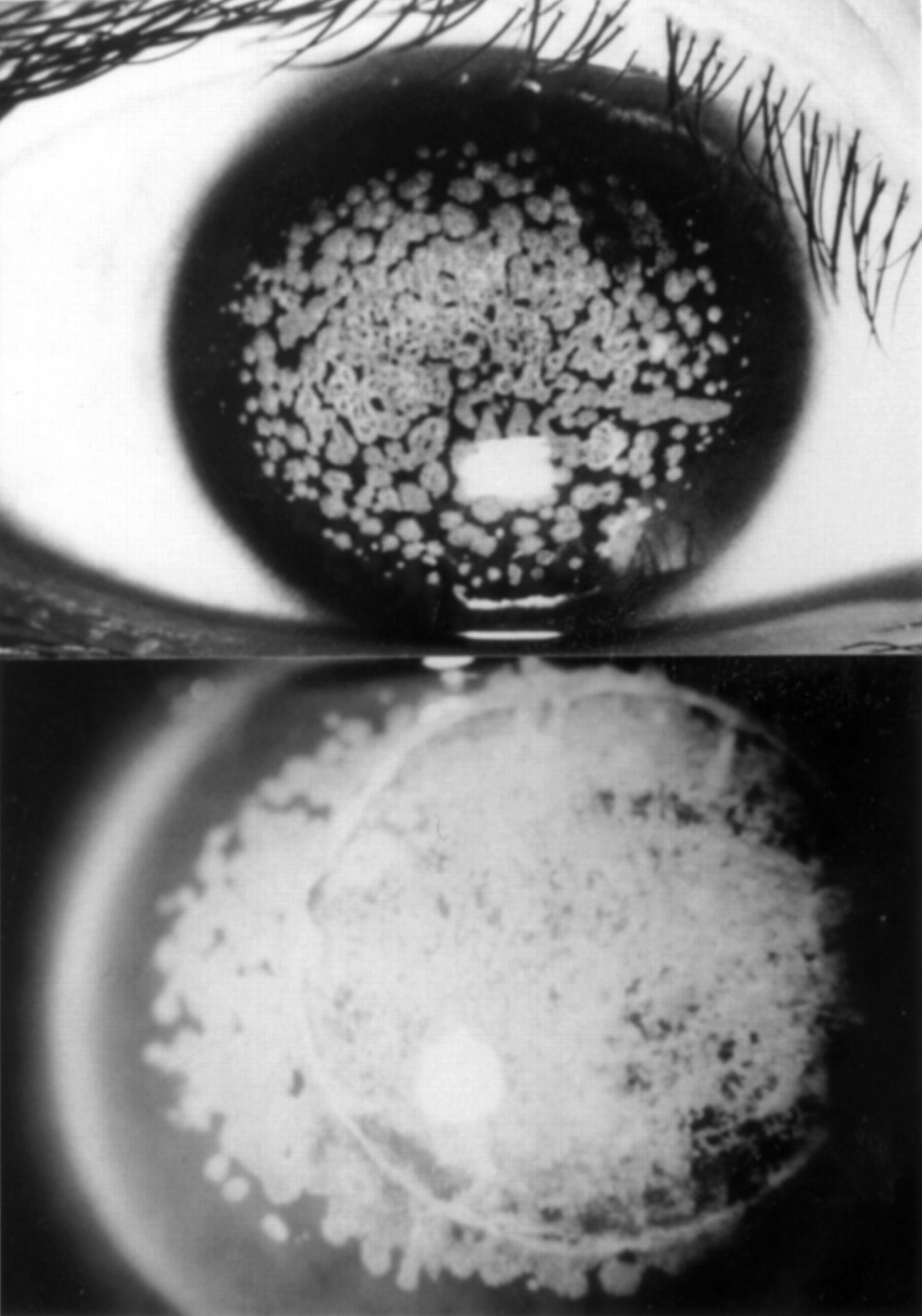
Slit lamp photographs of the cornea of case 2. Top, obtained preoperatively at the age of 13 years. Bottom, 14 years after the first surgery.
Case 3
A 59 year old Japanese woman noticed a decrease in visual acuity at the age of 10 years. She underwent superficial keratectomy in both eyes at the age of 16. After the operation, her visual acuity gradually decreased. She underwent lamellar keratoplasty in the right eye at the age of 21 and in the left eye at the age of 24. She visited Keio University Hospital at the age of 50 years in 1989 because of a decrease in visual acuity in both eyes due to severe recurrence after grafting. Visual acuity was reduced to finger counting in the right eye and 2/200 in the left eye (Fig 4, top left). She underwent penetrating keratoplasty in the right eye at the age of 50 years, and in the left eye at the age of 52 years. Paraffin sections of corneal specimens stained with Masson’s trichrome showed a red granular appearance. The deposits also stained moderately with Congo red, but did not exhibit birefringence and dichroism under polarised light. She was diagnosed as having a severe variant of juvenile GCD. Her grafts showed a recurrence of GCD after keratoplasty. Her best corrected visual acuity was 20/30 in the right eye and 20/50 in the left eye. Her parents were first cousins. Except for her brother, who suffered from the same eye disease, she said she had no relatives with impaired vision.

Slit lamp photograph of the corneas of recurrence. Untreated recipient corneas show dense subepithelial opacities in each case. Top left, case 3, 29 years after the second operation performed at the age of 21. Top right, case 4, 20 years after the first operation at the age of 25. Bottom left, case 5, 21 years after the second operation at the age of 53. Bottom right, daughter of case 5 at 48 years of age. Cornea shows only several granular opacities.
Case 4
A 58 year old Japanese man noticed a decrease in visual acuity in both eyes while still in elementary school. He underwent lamellar keratoplasty in both eyes at the age of 25 years. He visited Keio University Hospital at the age of 45 years in 1985 because of a decrease in visual acuity in both eyes—8/200 in the right eye and 4/200 in the left eye. His corneal grafts appeared to exhibit recurrence of GCD (Fig 4, top right). Penetrating keratoplasty was performed in the patient’s left eye when he was 45 years old, and the right eye 1 year later.
Histological study of the corneal specimen stained with Masson’s trichrome showed a red granular appearance. The corneal button also stained positively for Congo red with birefringence and dichroism observed under cross polarised light. These findings suggest a diagnosis of ACD. Examination of his grafts in 1997 showed recurrence of the disease. His best corrected visual acuity was 20/200 in the right eye, with the presence of macular degeneration, and 20/25 in the left eye. He had no corneal erosive episodes. He knew of no relatives who suffered from a decrease in vision. His grandparents were first cousins.
Case 5
A 74 year old Japanese woman was referred to Keio University Hospital in September 1997 for severe corneal opacities that caused a substantial impairment of vision in both eyes (Fig 4, bottom left). Her visual acuity was 20/200 in both eyes due to recurrence after grafting at the age of 53 years and the presence of senile cataract. Her parents reportedly first noticed whitish corneal opacities in both of her eyes when she was 4 years old. The patient noticed visual impairment at the age of 10 years. She underwent keratectomy in both eyes at the age of 18 years. Her visual acuity had decreased to finger counting in both eyes due to disease recurrence. She then underwent a penetrating keratoplasty in both eyes at the age of 53 years. She had no corneal erosive episodes. Her parents were first cousins. Examination of her daughter, aged 48 years (Fig 4, bottom right) and granddaughter, aged 22 years, showed a few granular opacities in the cornea.
MOLECULAR GENETIC ANALYSIS
Informed consent was obtained from all subjects. DNA was extracted from their peripheral blood leucocytes by standard methods. We investigated the five probands, six family members of case 1 (Fig 1, bottom right), including five affected individuals and one unaffected individual, and two affected relatives of case 5. Target DNA sequences within the BIGH3 gene were amplified by the polymerase chain reaction (PCR), according to the following thermocycling protocol: 5 minutes at 94°C; 35 cycles at 94°C for 1 minute, 55°C for 1 minute, and 72°C for 1 minute; and 72°C for 7 minutes. The primers used for PCR and direct sequencing analysis were EXON4F (5′-CCCCAG AGGCCATCCCTCCT-3′) and EXON4R (5′ - CCGGGCAGACGGAGGTCATC -3′).10 The PCR amplified products were sequenced by an automatic fluorimetric 377 DNA sequencer (Applied Biosystems) and a Prism DyeDeoxy Terminator cycle sequencing kit (Applied Biosystems) according to the manufacturer’s recommended protocols.
Results
MOLECULAR GENETIC ANALYSIS
Direct sequencing confirmed a homozygous G → A transition (Arg → His) at codon 124 of the BIGH3 gene in all five patients. In the family of case 1, the proband (IV-10) showed a homozygous G → A transition, while other affected family members were heterozygous for the mutation (Fig 1, bottom right and Fig 5). The proband’s unaffected sister (IV-11 in Fig 1, bottom right) did not harbour the mutation. In the family of case 5, her daughter and granddaughter showed a heterozygous G → A transition.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Direct sequencing analysis of the BIGH3 gene (exon 4) with an automated fluorimetric sequencer. Top, case 1. A homozygous G → A nucleotide substitution was detected at the second position of codon 124 (CGC) (arrow). Bottom, her mother. A heterozygous G → A nucleotide substitution was apparent at the second position of codon 124 (CGC). Two waves of G (black) and A (green) coexist within one peak (arrow). Top line shows normal DNA sequence.
Discussion
Four of our five severely affected probands with the homozygous R124H mutation, all of whom were the offspring of consanguineous parents, underwent keratoplasty before the age of 16 because of the early onset and rapid progression of the disease. Corneal deposits reappeared less than a year after surgery and the disease showed unusually rapid progression. These homozygous patients therefore required additional keratoplasties. However, recurrent disease was not as progressive after additional keratoplasties were performed when they were in their 40s or 50s. The brother, parents, and affected grandparents of case 1 were heterozygous for the R124H mutation. Her unaffected sister lacked this mutation. Keratoepithelin is a secretory protein and is expressed on the extracellular surface of the corneal epithelium.14 The mutant homozygous offspring developed a greater number of corneal deposits than her heterozygous parents with this autosomal dominant disease. This observation suggests that the phenotype of the corneal manifestations depends on the dose effect of the mutant gene. Previous reports have described severely affected patients with early onset GCD who were the offspring of consanguineous parents, although these reports did not fully describe the condition of the corneas of the parents.2 5 8 9 In only a few families were both parents reportedly affected.6 7Because most heterozygous parents with this corneal dystrophy do not have visual symptoms until the fifth decade of life, diagnosis cannot be based on subjective symptoms, but rather requires a slit lamp examination.
The severe form of juvenile GCD has mainly been reported in Japan.2 9 15-18 The appearance of the cornea in these reported cases in the patient’s first decade of life resembled the cornea of cases 1 and 2 in this study. In most cases, the patients’ parents were consanguineous. Two or three generations ago, consanguineous marriages were not rare in Japan. Three of our five patients were over the age of 50 years. In a previous mutational analysis of the BIGH3 gene in Japanese patients with stromal corneal dystrophies, only a homozygous R124H mutation was observed in patients with ACD, and not in those with lattice corneal dystrophy type 1 (LCD1).11 This observation reflects the fact that most heterozygous individuals with the R124H mutation do not have visual symptoms in the second or third decade of life. Thus, because individuals with ACD have no visual symptoms until at least their 40s, a consanguineous marriage is a possibility.
ACD exhibits clinical and histological features of both GCD and LCD1.19-22 Previous reports of individuals with GCD have described amyloid deposits in the corneas of older patients,23 24 and it has been suggested that amyloid deposits develop late in the course of GCD.20 21 24Amyloid deposits were not detected in corneal specimens obtained from case 1 at 10 and 20 years of age. Case 1 showed similar clinical and histological features to previously described young patients with superficial variant GCD,2 7-9 which is consistent with the theory that amyloid deposits rarely appear in the first or second decade of life in patients with ACD.
Molecular genetic analysis has shown that the R124H mutation cosegregates with the phenotype of ACD in the heterozygous state, whereas it causes a superficial juvenile variant form in the homozygous state. Our results indicate that direct corneal examination and histological study may be insufficient to establish a definite diagnosis of these corneal conditions and that analysis of mutations of the keratoepithelin gene may be required.