Article Text

Abstract
BACKGROUND/AIMS Tissue inhibitor of metalloproteinases-3 (TIMP-3) is normally synthesised by the retinal pigment epithelium (RPE) and deposited in Bruch’s membrane. Mutations in the TIMP3 gene cause Sorsby’s fundus dystrophy (SFD), which is characterised by thickening of Bruch’s membrane, choroidal neovascularisation, and photoreceptor degeneration. To elucidate the role of TIMP-3 in human retinal degenerative diseases, we immunolocalised TIMP-3 in eyes with SFD caused by the Ser-181-Cys TIMP3 gene mutation or retinitis pigmentosa (RP; not caused byTIMP3 mutations).
METHODS Standard light microscopic immunocytochemistry, including antigen retrieval, was used to localise TIMP-3 in paraffin sections of human eyes: two with SFD, three with different genetic forms of RP, and two normal.
RESULTS In the SFD eyes, the thickened Bruch’s membrane was strongly TIMP-3 positive except where RPE cells had degenerated. Similarly, in the RP eyes, Bruch’s membrane was TIMP-3 positive except where RPE cells were lost, consistent with ongoing RPE mediated turnover of TIMP-3 in this region. In areas of total photoreceptor loss, migrated RPE cells formed cuffs around blood vessels in the RP retinas. Thick, TIMP-3 positive extracellular matrix (ECM) deposits associated with the migrated RPE cells occluded some vascular lumina, correlating with the observed loss of inner retinal neurons in RP.
CONCLUSIONS TIMP-3 is a component of the increased ECM sequestered in Bruch’s membrane in SFD. Further information is needed on normal TIMP-3/ECM interactions in Bruch’s membrane and the effect of mutant TIMP-3 on this process. The finding of TIMP-3 accumulations in retinas with RP not caused by TIMP-3 mutations emphasises the importance of ECM remodelling in normal and diseased human eyes.
- tissue inhibitor of metalloproteinases-3
- Sorsby’s fundus dystrophy
- retinitis pigmentosa
- inherited retinal diseases
Statistics from Altmetric.com
- tissue inhibitor of metalloproteinases-3
- Sorsby’s fundus dystrophy
- retinitis pigmentosa
- inherited retinal diseases
In Sorsby’s fundus dystrophy (SFD), an autosomal dominant retinal degeneration,1 extracellular deposits accumulate in Bruch’s membrane, the five layered sheet of connective tissue that separates the retinal pigment epithelium (RPE) from its blood supply, the choriocapillaris.2 It was suggested3 that these abnormal sub-RPE deposits interfere with transport of essential molecules from the choriocapillaris to the RPE, leading to dysfunction and death of the RPE cells and retinal photoreceptors. A serious complication of SFD is invasion of the thickened Bruch’s membrane by newly formed, thin walled vessels derived from the choriocapillaris. These new vessels can grow into the subretinal space, causing exudative detachment of the RPE and photoreceptor demise.4
Patients with SFD have mutations in exon 5 of the gene for TIMP-3 (tissue inhibitor of metalloproteinases-3).5-12 TIMP-3 is a member of a family of matrix metalloproteinase (MMP) inhibitors13-15 thought to act as local regulators of matrix degradation by the MMPs. Recent studies have localised TIMP-3 protein to Bruch’s membrane16-18 and TIMP-3 mRNA to RPE cells,17 19-21 and there is general consensus that the TIMP-3 in Bruch’s membrane is synthesised and secreted by the RPE. It was suggested5 6 22 that TIMP-3 normally functions for maintenance of the extracellular matrix (ECM) in Bruch’s membrane and that a mutant TIMP3 gene product may lead to accumulated sub-RPE deposits in SFD by interfering with the normal balance between ECM deposition and degradation.
Although many questions remain about the role of mutant TIMP-3 in the pathology of SFD, no animal models are yet available and the definitive histopathological study of SFD eyes2 was performed before causative TIMP-3 mutations were identified. Using a recently developed monoclonal antibody (mAb) against human TIMP-3,16 we have documented the distribution of TIMP-3 in human eyes with SFD caused by the Ser-181-Cys TIMP-3 mutation.
We also wished to determine if TIMP-3 localisation in SFD was unique or might also be present in a dominant form of retinitis pigmentosa (RP) not caused by a TIMP-3 mutation but having sub-RPE deposits resembling those in SFD.23 Finally, RPE abnormalities secondary to photoreceptor death are common in other forms of RP,23-25including those caused by mutations in photoreceptor specific genes such as rhodopsin. These RPE abnormalities include cellular detachment from Bruch’s membrane and migration to perivascular sites in the inner retina,25 suggesting that TIMP-3 localisation might also be abnormal. To answer these questions, we extended our TIMP-3 immunolocalisation studies to RP retinas with documented abnormalities in Bruch’s membrane and the RPE.
Materials and methods
DESCRIPTION OF DONOR EYES
The eyes of a 77 year old woman with SFD caused by theSer-181-Cys TIMP3 gene mutation were obtained post mortem. This patient donor was a member of family 13 in table 2 of a recent report,10 and was patient IV-4 in pedigree plate IV of the original report by Sorsby and Mason1 and patient IV-4 in the pedigree in figure 1 of Capon et al.26 The retinal histopathology of the first cousin of our donor has been reported.2 Our SFD donor had developed poor night vision in her early 30s and by her early 40s, had distorted vision in her left eye. Her visual acuities decreased to hand movements after choroidal neovascularisation and disciform scars developed in both maculas. At age 57, a retinal detachment in the left eye was treated by scleral buckling, and at age 70, cataract surgery was performed on the right eye.
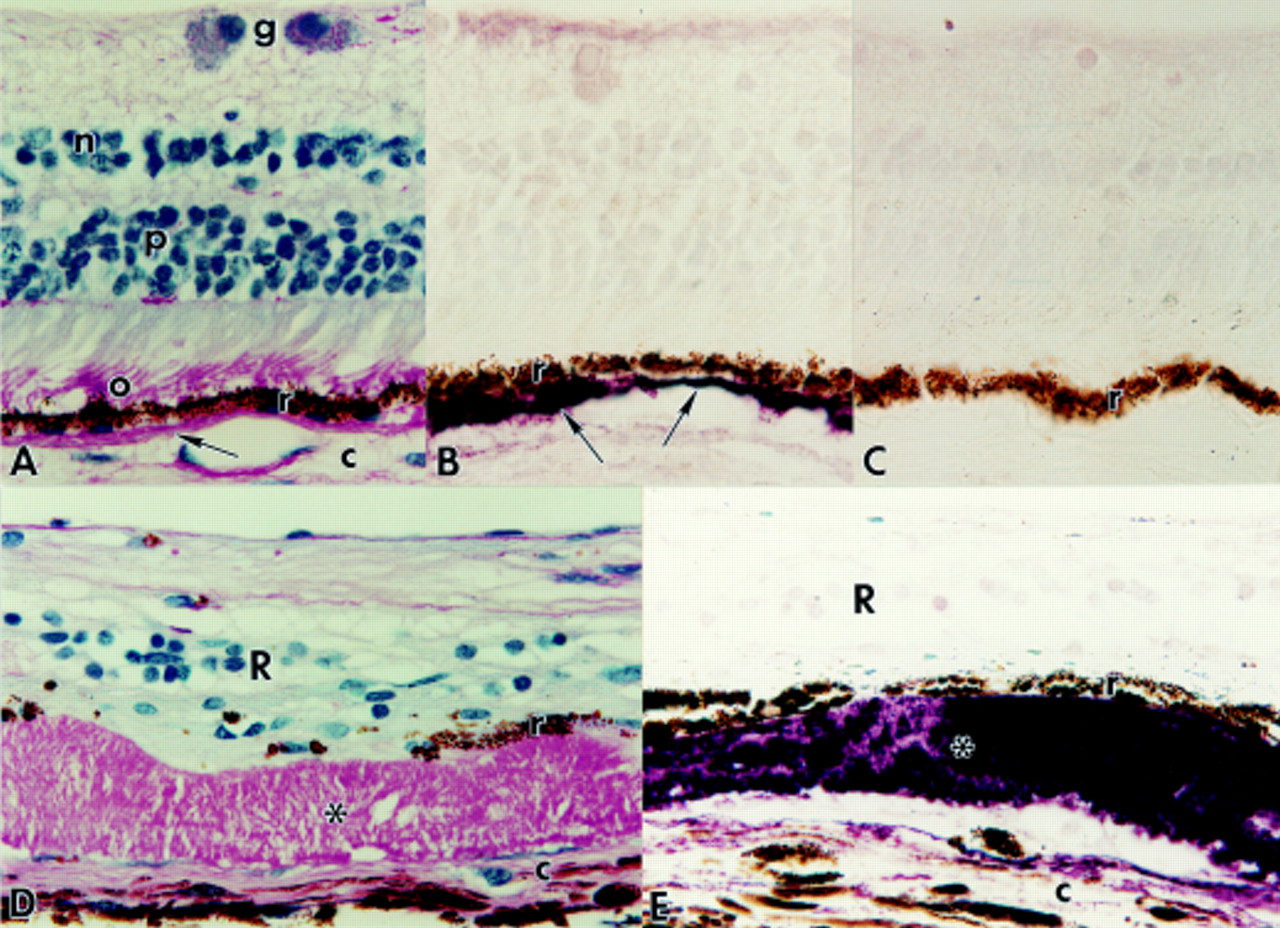
Paraffin sections of normal human eyes (A, B, and C) and of eyes with Sorsby’s fundus dystrophy (SFD: D and E). Magnification, ×625. (A) Normal retina stained by PAS and haematoxylin. Bruch’s membrane (arrow) is PAS positive. o=PAS positive photoreceptor outer segments; r=retinal pigment epithelium (RPE); c=choroid; p=photoreceptor nuclei; n=inner nuclear layer; g=ganglion cell layer. (B) Normal retina immunolabelled with anti-TIMP-3. Note strong reactivity (purple reaction product) of Bruch’s membrane (arrows), which is somewhat thickened on the left side of the figure. r=RPE. (C) Normal retina processed for immunocytochemistry with secondary antibody alone. Only weak, non-specific labelling of scattered nuclei in the retina is observed. r=RPE. (D) SFD retina stained by PAS and haematoxylin. Note thick layer of PAS positive deposits (*) between the RPE (r) and choroid (c). The retina (R) is severely degenerate. (E) SFD retina immunolabelled with anti-TIMP-3. Note strong reactivity (purple) of the sub-RPE deposits (*). r=RPE; c=choroid; R=retina.
Samples were taken from areas in the periphery of both retinas, not including areas involved in the previous detachment. The retinal samples were processed in paraffin, while the maculas and remainder of the retinas were processed for tissue culture. Samples from the eyes of three patient donors with RP, as well as two normal donors with no history of ophthalmic disease (Table 1) were also processed in paraffin. The retinal histopathology of RP donors FFB-356 and FFB-316 has been reported in detail.23 27 Donor FFB-356 had dominant RP of unknown genotype but mutations in TIMP3, rhodopsin, and rds/peripherin were excluded.23 Donor FFB-316 had dominant RP caused by a rhodopsin mutation,27 and donor FFB-516 had advanced dominant RP of unknown genotype (Table 1).
Characteristics of human retinas used in study
ANTIBODIES
Immunocytochemistry was performed with a mouse mAb to human TIMP-3 prepared against a C-terminal domain synthetic peptide containing residues 170–188 (10 μg/ml, clone 136–13H4; from Dr K Iwata, Fuji Chemical Industries, Ltd, Takaoka, Japan).16 Western blotting verified that this mAb recognises TIMP-3 but not TIMP-1 or TIMP-2.16 To document new vessel growth in the SFD eyes, sections were processed for immunocytochemistry using mouse mAb anti-factor VIII (von Willebrand factor) (1:200; Dako Corporation, Carpinteria, CA, USA). The elastin sublamina of Bruch’s membrane was labelled with anti-elastin (1:50; Elastin Products, Inc, Owensville, MO, USA)
TISSUE PROCESSING
Paraffin sections (8 μm) were prepared from the eyes after fixation for several days in formalin or 4% paraformaldehyde in 0.13 M phosphate buffer, pH 7.3, with or without added 0.5% glutaraldehyde (Table 1). The paraffin sections were deparaffinised in xylene, rehydrated through an alcohol series, and processed for antigen retrieval,28 including microwave treatment for 4 minutes in citrate buffer, pH 3.5, followed by incubation for 20 minutes in 2% β mercaptoethanol, 1% sodium borohydride and 10% methanol in citrate buffer, pH 2.8. The sections were incubated overnight in anti-TIMP-3, anti-elastin, or anti-factor VIII at 4°C and processed further using a Vectastain avidin-biotin-complex kit (Vector Laboratories, Burlingame, CA, USA). Control sections were processed in the same way with omission of primary antibody, or with anti-TIMP-3 preadsorbed with TIMP-3 peptide (10–50 μg/ml). Additional sections were stained by the periodic acid Schiff (PAS) method with haematoxylin counterstaining.
Results
In the normal eyes, Bruch’s membrane was consistently PAS positive and ranged in thickness from 3 to 8 μm (Fig 1A). The central sublamina of Bruch’s membrane was strongly immunoreactive for elastin. Bruch’s membrane and occasional drusen showed strong, specific TIMP-3 labelling (Fig 1B). Endothelial cells of the retinal and choroidal vessels were immunoreactive with anti-factor VIII but, as noted previously,16 no specific TIMP-3 labelling of the retinal blood vessels was detected in the normal eyes after paraffin embedding and antigen retrieval. Control sections of normal and diseased eyes processed for immunocytochemistry with omission of primary antibody or with anti-TIMP-3 preadsorbed with TIMP-3 peptide had no specific labelling although scattered retinal nuclei showed weak, non-specific reactivity (Fig 1C).
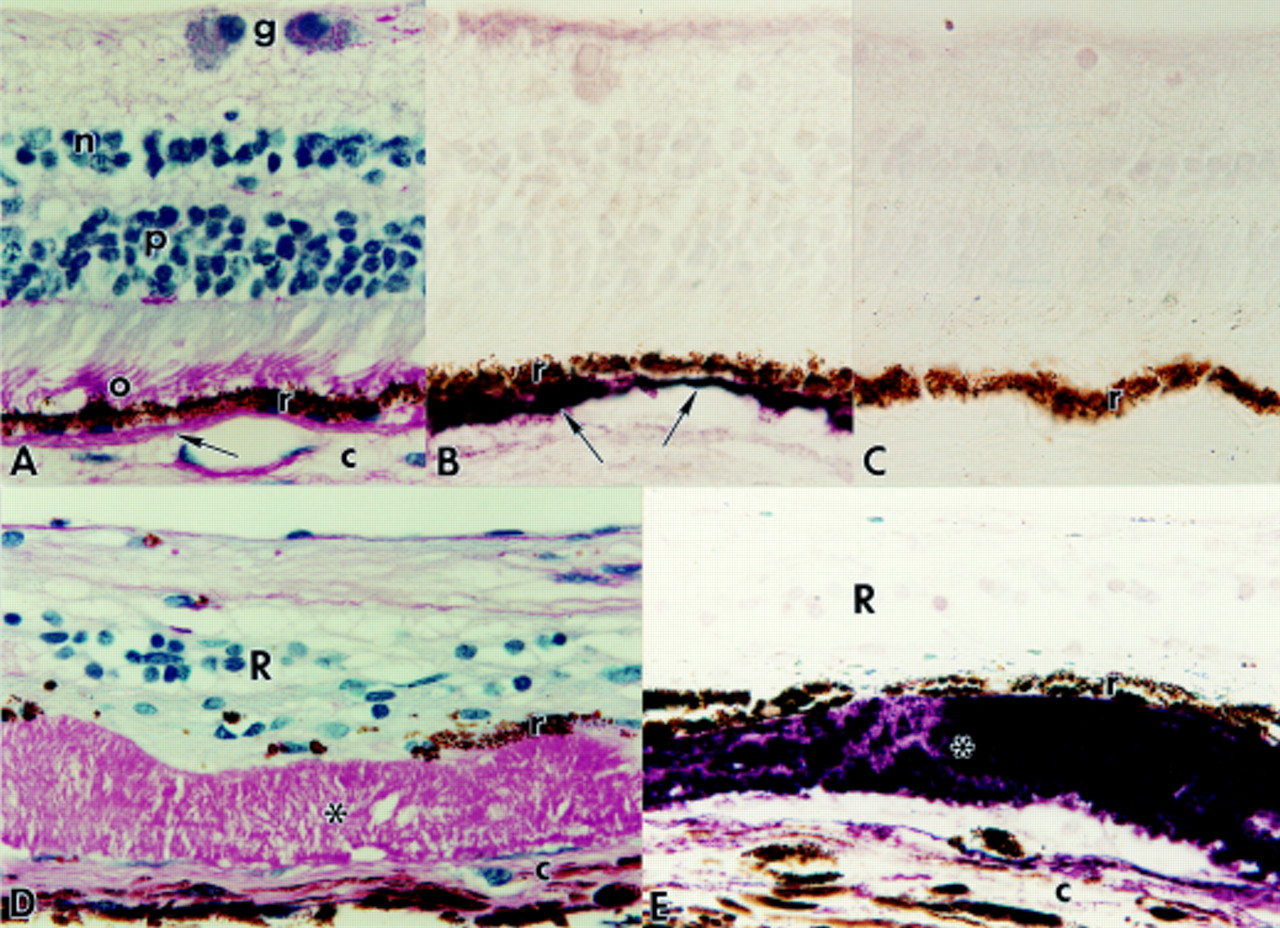
The SFD eyes showed advanced pathology, including loss of virtually all retinal photoreceptors and gliosis of the remaining retinal layers. Bruch’s membrane was markedly thickened with extracellular PAS positive deposits that ranged in thickness from 15 to 60 μm (Fig 1D). The deposits, which were located between the RPE and elastin sublayer of Bruch’s membrane, were strongly TIMP-3 positive throughout their thickness where the RPE cells were retained (Fig 1E). No TIMP-3 reactivity was found in the cytoplasm of the melanin containing RPE cells, which were thinned but usually retained as a monolayer next to the deposits. In occasional areas where RPE cells had been lost, the adjacent Bruch’s membrane deposits remained PAS positive (Fig 2A) but had no TIMP-3 immunoreactivity (Fig 2B). The fate of the missing RPE cells was not apparent; RPE cell migration to perivascular sites in the inner retina was not observed, as found frequently in RP retinas (Figs 2C and D).

{kind=link}
{kind=link}
Paraffin sections of retinas with Sorsby’s fundus dystrophy (SFD, A and B) and retinitis pigmentosa (RP, C and D). Magnification, ×625. (A) SFD retina stained by PAS and haematoxylin. The thick subretinal pigment epithelium (RPE) deposits (*) are variable in thickness but PAS positive throughout. r=RPE; c=choroid; R=degenerate retina. (B) SFD retina section adjacent to (A) immunolabelled (purple reaction product) with anti-TIMP-3. Note strong TIMP-3 reactivity of sub-RPE deposits (black *) adjacent to RPE cells (r) but absence of TIMP-3 reactivity in area (blue *) where RPE cells have been lost. c=choroid; R=retina. (C) RP retina (FFB-316) stained by PAS and haematoxylin, illustrating bone spicule pigment deposits formed by migrated RPE cells (r) that surround inner retinal blood vessels. The matrix (*) deposited by the perivascular layer of RPE cells is strongly PAS positive and occludes the retinal blood vessels. c=choroid; R=degenerate retina. (D) Section of RP retina adjacent to (C) processed for immunocytochemistry with anti-TIMP-3 (purple reaction product). Note strong labelling of matrix deposits (*) that occlude the retinal blood vessels. Arrow indicates TIMP-3 positive region of Bruch’s membrane adjacent to RPE cells (r). Note loss of TIMP-3 reactivity in region of Bruch’s membrane (blue *) where RPE cells have migrated away. c=choroid; R=retina.
Sections of the SFD eyes processed with anti-factor VIII showed specific labelling of endothelial cells in retinal blood vessels, which were reduced in number and calibre, consistent with advanced retinal degeneration. Specific labelling with anti-factor VIII was also present in endothelial cells of the large choroidal vessels and the choriocapillaris, which was atrophic, consistent with severe photoreceptor loss. In the peripheral retinal regions examined, no choroidal vessels were observed to pass through Bruch’s membrane or to occupy the subretinal space, consistent with the SFD patient’s history of choroidal neovascularisation limited to the macular regions.
As found in the SFD eyes, the 80 year old RP eye of unknown genotype (but documented not to have a TIMP3 gene mutation)23 had extremely thick (∼50 μm) deposits internal to the elastin sublayer of Bruch’s membrane. These deposits, which were PAS positive throughout, were strongly TIMP-3 positive adjacent to surviving RPE cells but less TIMP-3 reactive where RPE cells had degenerated.
In the other RP retinas, Bruch’s membrane was PAS positive and well labelled with anti-TIMP-3 where RPE cells were present (Fig 2D). The 68 and 81 year old RP retinas showed normal aging changes, including slight thickening of Bruch’s membrane, and had a few TIMP-3 positive drusen internal to the elastin sublamina. In addition, large areas in the RP retinas had total loss of photoreceptors and gross deposits of bone spicule pigment. In these areas, the RPE cells had migrated inward from Bruch’s membrane and formed perivascular cuffs in the inner retina (Fig 2C). As in the SFD eyes, the regions of Bruch’s membrane where RPE cells were no longer in situ had lost TIMP-3 reactivity (Fig2D) but remained PAS positive.
The inner retinal vessels surrounded by migrated RPE cells (Fig 2C) had prominent accumulations of PAS positive ECM between the RPE and endothelial cells, the latter identified by labelling with anti-factor VIII. In a previous electron microscopic study,25 the perivascular deposits of ECM were found to resemble Bruch’s membrane in situ, having its characteristic five sublayers with a central layer of elastin not normally found around retinal blood vessels. As noted in normal Bruch’s membrane, the perivascular deposits of ECM associated with the RPE cells were strongly TIMP-3 positive (Fig 2D). In some vessels, the thick deposits of TIMP-3 reactive ECM occluded the lumina (Fig 2D).
Discussion
The turnover of ECM in many tissues results from a balance between matrix synthesis and degradation, the latter due in part to proteolytic action of MMPs that is opposed by their inhibitors, the TIMP family of proteins. The role of TIMP-2 in activation of progelatinase A29 and the affinity of TIMP-3 for the ECM13suggest that TIMP interactions with MMPs are more complex than simple enzyme inhibitor reactions. In particular, TIMP-3 binding to the ECM is poorly understood, as are the implications of this binding for MMP inhibition and matrix turnover.
As a first step to elucidate the underlying disease mechanisms in SFD, we examined TIMP-3 distribution in a pair of SFD eyes and found that the thick Bruch’s membrane deposits were strongly immunoreactive for TIMP-3. To our knowledge, this represents the first documentation of TIMP-3 localisation in eyes with SFD. Although it has been suggested5 that TIMP-3 mutations affect the function of TIMP-3 as a MMP inhibitor, the accumulation of abnormal matrix deposits in Bruch’s membrane is probably not due simply to loss of function in mutant TIMP-3 because the amount of ECM is markedly increased, not decreased, in SFD. There is evidence that the thickened Bruch’s membrane in SFD differs in composition from that in normal eyes,2 and it is possible that mutant TIMP-3has abnormal matrix binding properties. However, preliminary data (Anand-Apte et al, unpublished) indicate that the antibodies used in the present study do not distinguish between normal and mutant (Ser-181-Cys) TIMP-3, so it is unclear if mutant TIMP-3 contributes to the strong immunolabelling of the sub-RPE deposits in the SFD eyes found in our study. Our findings do not show any evidence of reduced expression of TIMP-3in SFD.
TIMP-3 reactivity was absent in the RPE-free regions of Bruch’s membrane in both the SFD and RP eyes. This observation is consistent with more rapid turnover of TIMP-3 in Bruch’s membrane relative to other matrix components—for example, those stained by PAS. Although our results emphasise the role of RPE in the turnover of TIMP-3 in Bruch’s membrane, they do not reveal if there is a difference in turnover of mutant versus normal TIMP-3 in SFD.
The sequence of events leading from a TIMP3 mutation to the characteristic choroidal neovascularisation in SFD4 5is undoubtedly complex and presently unknown. Choroidal neovascularisation occurs in any condition in which Bruch’s membrane becomes thickened—for example, age related macular degeneration (ARMD), so this complication of SFD may not be directly attributable to the primary gene defect. Although, TIMP-3 is anti-angiogenic both in vitro and in vivo,30 it has not been shown that this effect is altered by the TIMP3 mutations associated with SFD. It would be desirable to assess the distribution of TIMP-3 in areas of choroidal neovascularisation in SFD eyes, but unfortunately such vessels could not be identified in the SFD retinal samples available for the present study. Additional work is needed to determine how mutations in the TIMP3 gene lead to the characteristic ocular phenotype of SFD.
We know of no evidence that TIMP3 mutations cause any form of RP or other retinal diseases aside from SFD.31 32 However, because sub-RPE deposits and RPE abnormalities are found in RP retinas,23-25 it was also of interest to document the localisation of TIMP-3 in this disease. The finding of TIMP-3 reactive ECM in deposits of bone spicule pigment may correlate with the increase in TIMP-3 mRNA noted in RP retinas.33 However, we obtained no evidence for immunoreactive TIMP-3 in photoreceptors or ganglion cells in the RP retinas, as suggested by previous in situ hybridisation34and immunocytochemical21 studies. The reason(s) for this apparent discrepancy are unknown. TIMP-3 immunoreactivity outside of Bruch’s membrane in the RP retinas was found only in regions of total photoreceptor loss where the RPE cells had migrated to perivascular sites in the inner retina. The observation of TIMP-3 reactive ECM deposits occluding the lumina of the retinal blood vessels may help to explain the loss of inner retinal neurons observed in RP.35 36 It is possible that local inhibition of MMP activity by increased perivascular TIMP-3 leads to ECM accumulations that diminish or block blood flow,37 resulting in hypoxic death of inner retinal neurons.
Because TIMP-3 is a component of thick ECM deposits in Bruch’s membrane in SFD, ARMD,18 and at least one form of RP23 it is essential to elucidate the mechanism of TIMP-3 binding in this region. In SFD, several different point mutations are known to occur in TIMP-3, while in ARMD and RP, none have been reported. What then accounts for this common pattern of TIMP-3 accumulation in Bruch’s membrane? Perhaps mutant TIMP-3in SFD undergoes slower turnover in Bruch’s membrane, although results from the present study suggest that TIMP-3 undergoes more rapid turnover than the components of Bruch’s membrane stained by PAS. In diseases such as ARMD and RP not caused by TIMP-3mutations, the regulation of TIMP3 gene expression may be altered or TIMP-3 may bind to abnormal molecules accumulated in Bruch’s membrane, resulting in a net increase in ECM resembling that in SFD. Additional information is needed on the components of Bruch’s membrane bound by both wild type and mutant TIMP-3 in order to understand the underlying disease mechanisms in the various human retinal dystrophies.
Acknowledgments
Supported by The Foundation Fighting Blindness (Hunt Valley, MD) (AHM and ACB); Research to Prevent Blindness, Inc (New York, NY) (AHM); and by NIH grants AR44436 (SSA), EY01311 (AHM), and EY01730 (AHM). Legal requirements for use of human donor postmortem tissues were met (University of Washington Human Subjects Approval No 27–0060-E, dated 16 December 1996). Human donor eyes were provided by the University of Washington Medical Center and Lions Eye Bank, supported by the Washington and Northern Idaho Lions Sight Conservation Foundation, Seattle, WA, USA.
Proprietary interests: None
The authors are grateful to Ms J Chang, Ms I Klock, Ms C Ulness, Ms M Wilhyde, and Ms R Small for technical assistance, to Dr K Iwata of Fuji Chemical Co Ltd for providing the anti-TIMP-3, to Dr Z-Y Li for immunocytochemistry advice, and to Drs K Evans and SG Jacobson for review of the manuscript.