Article Text

Abstract
AIM X linked retinitis pigmentosa (XLRP) has two genetic loci known as “RP2” and “RP3”. Clinical features reported to differentiate RP2 from RP3 include a higher prevalence of myopia and primary cone dysfunction in RP2, and late onset night blindness and tapetal reflex in RP3. Members from 14 XLRP families were examined in an attempt to verify these differences.
METHODS 16 affected males and 37 females from 14 XLRP families assigned as either RP2 or RP3 by haplotype analysis and/or by heterogeneity analysis were examined. Members of all 14 families who were willing to participate but unavailable for examination were contacted and detailed interviews carried out.
RESULTS No clear phenotypic differences were found that could be used to reliably differentiate RP2 from RP3 with respect to myopia and onset of night blindness. The tapetal reflex was also found to be present in carriers of both RP2 and RP3.
CONCLUSIONS XLRP is a heterogeneous class of rod degenerative disorders with no clear phenotypic differentiation between the two genetic loci RP2 and RP3. There is a continuum of clinical presentations which can be seen in both RP2 and RP3, but the features within a given family tend to be consistent. However, interfamilial variability is prevalent leading to a wide range of clinical presentations and more than one abnormal allele at each gene locus cannot be excluded.
- RP phenotypes
- X linked retinitis pigmentosa
Statistics from Altmetric.com
Retinitis pigmentosa (RP) is a well known variably progressive hereditary retinal disorder in which all Mendelian modes of inheritance have been described.1 2 It primarily alters rod photoreceptor function as well as affecting the retinal pigment epithelium.2 X linked recessive retinitis pigmentosa (XLRP) is genetically and phenotypically heterogeneous for both affected males and carrier females.3-8 Two distinct genetic loci have been described and assigned as RP2 (corresponding to the Xp11.2–3 locus) and RP3 (corresponding to the Xp2.1 region).1 7 9 10 RP2 and RP3 phenotypes have been distinguished on the basis of tapetal reflex seen in some carrier females in RP3 families,1 6 11 12 but there have been no clear distinctions between the two gene loci and clinical presentation. Recently, attempts have been made to correlate phenotypes with genetic locus. Souied and co-workers have previously described two XLRP clinical profiles and recently reported a third “partially dominant” XLRP profile.13 The first profile shows an earlier onset of myopia in RP2 hemizygotes and the second shows a later onset of night blindness in RP3 hemizygotes.7 The recently reported third profile shows delayed onset in affected males with severe expression in carrier females and has been mapped to the RP3 locus.13 Jacobson and colleagues subsequently reported one family of the RP2 genotype in which the phenotype was initially expressed as a cone photoreceptor dysfunction with subsequent involvement of both rod and cone systems as the disease progressed.9
Questions have arisen as to whether these reports describe common but unrecognised findings and whether other differences exist between the RP2 and RP3 genotypes which have not been recognised. There is currently no widespread agreement as to whether such clinical differences exist between the phenotypes produced by the two loci, and the possibility of more than one abnormal allele at each locus has not been excluded.1
We undertook examination and review of males and females belonging to British XLRP families who had prior genetic studies classifying them as either RP2 or RP3 in an attempt to document phenotypic characteristics and variability.
Patients and methods
A list of 26 British families with XLRP classified as either RP2 or RP3 by genetic analysis was obtained from the Institute of Ophthalmology in London and the MRC Human Genetics Unit in Edinburgh (Table 1). Heterogeneity analysis as described by Teagueet al4 was carried out at the MRC Human Genetics Unit, Edinburgh, as well as haplotype analysis using genetic markers embracing the region of Xp containing both loci. Briefly, heterogeneity analysis is a method by which data obtained via haplotype analysis is pooled and a statistical estimate of the genotype of the family is obtained.
Details of families participating in study
Haplotype analysis was also carried out at the Institute of Ophthalmology, London, on most of the families used in the study. Families for which recombination events clearly positioned the XLRP locus proximal to DX57 (Xp11.3) or between CYBB and DX57 (Xp21.1-p11.3) were classified as RP2 or RP3 accordingly (Hardcastle AJ, unpublished data 1997).
The 26 families were contacted and asked to participate in a survey of XLRP, 18 families responded and agreed to participate. One family was subsequently excluded when a carrier female was examined and noted to have features typical of the choroideraemia carrier state, three other families were excluded because definitive classification as RP2 or RP3 could not be verified.10 Therefore a total of 14 families are included in this report.
At least one affected male and one carrier female were examined from each of the 14 participating families and details regarding earlier examinations and ocular history were obtained on all other family members when possible. During the current review, at least one carrier female was examined from each family and at least one affected male from each of the 14 families was currently examined or had previously been examined by one of the authors. Therefore, a total of 16 affected males and 37 females were examined and clinical details were obtained for unavailable family members to investigate any intrafamilial patterns with respect to age of onset of night blindness and degree of cone dysfunction. Thirty three of the 37 females examined were carriers for the XLRP gene by genetic determination.
The age of those patients examined ranged from 21 to 65 years with average age of 35 years. All patients examined underwent complete ocular examination including measurement of best corrected Snellen visual acuity, intraocular pressure measurement by applanation tonometry, slit lamp biomicroscopy, and dilated fundus examination. Electroretinography, visual field testing, and fundus fluorescein angiography were performed as clinically indicated. Colour vision testing was performed when possible utilising standard Ishihara colour plates administered in a standard manner.
We set out to obtain answers to several questions by examination, record review, or by direct questioning with regard to the affected males and heterozygous females in these 14 families. In the affected males, we queried the presence, degree, and age of onset of night vision loss; onset, presence, and degree of myopia; age when glasses were first required; ability to read fine print and discern colours; and age when difficulties with these tasks were first noted. In the carrier females, we queried the presence of any ocular complaints especially with regard to night vision, presence of myopic refractive error, and presence of fundus abnormalities. Several of the females had fundus fluorescein angiography and electrophysiological testing before the genetic determination of their carrier state, in an attempt to clarify their carrier status.12
Results
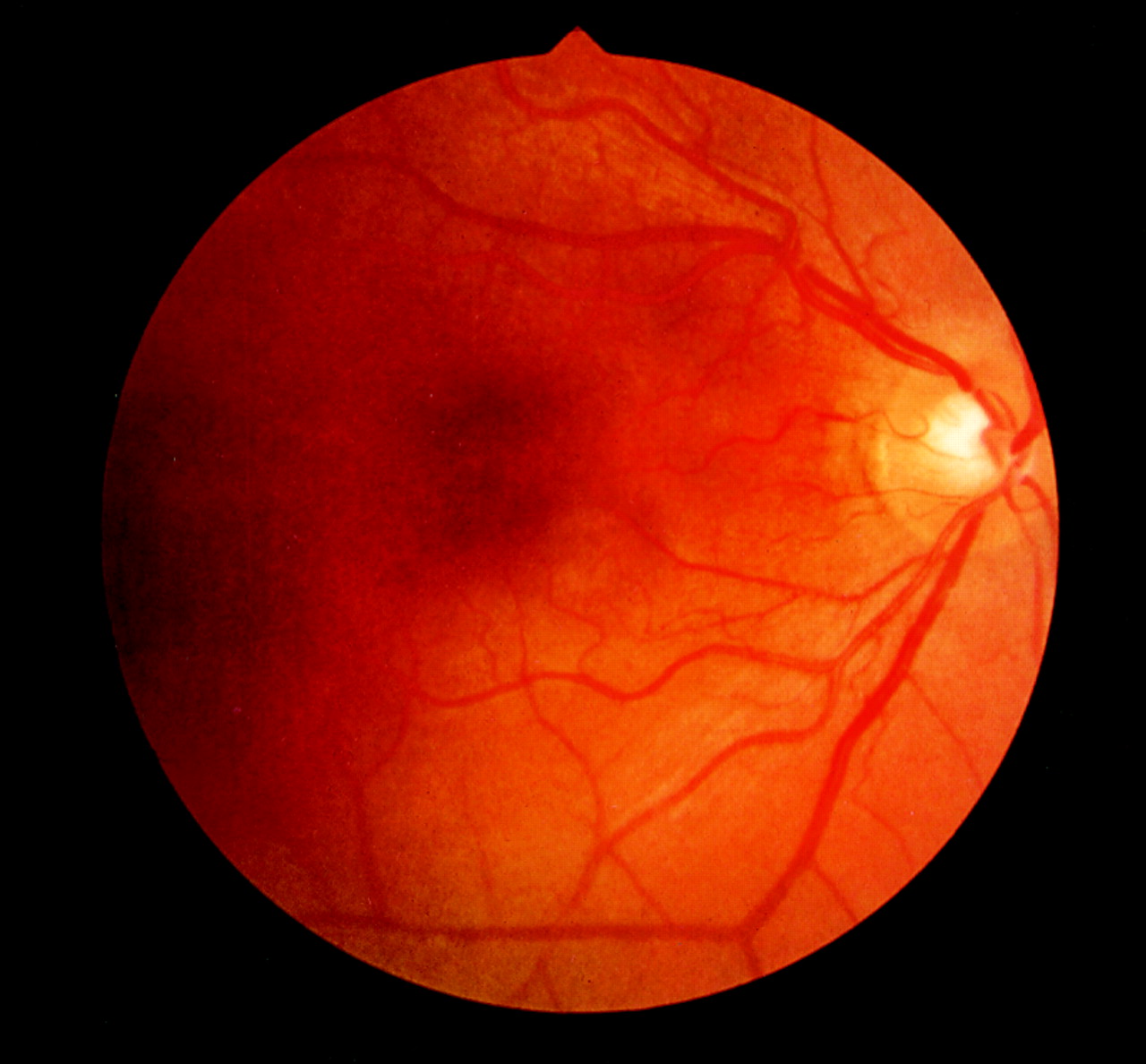
Results are summarised in Table 1. Three of the 14 families were classified as RP2 and 11 as RP3, this greater number of RP3 families is consistent with earlier reports that have found RP3 as the more common form of XLRP.4 8 Visual acuity in all affected males over the age of 50 years was poor, ranging from 6/60 to perception of light (PL) with evidence of severely limited visual fields. Central visual acuity was generally preserved in the younger affected males though they also had severely limited visual fields. All affected males had characteristic findings of RP on fundus examination (Fig 1). Best corrected Snellen visual acuity ranged from 6/5 to 6/9 with mean of 6/6 in the females examined. Of the 14 families reviewed, there was one family (family 3) with affected males with myopia greater than −5.00 dioptres but no carrier females with this degree of myopia.14 However, seven of the 14 families had a history of affected males with low myopia (below −5.00 dioptres), six of these families were RP3 and one was RP2. The remaining six families had no history of myopic refractive errors. The carrier females in family 14 also have a strong history of low myopia (RP3) but no other significant history of refractive error was obtained regarding the carriers. None of the individuals with myopia recalled the need for glasses before the age of 10 years.

Left fundus photograph of hemizygotic male from family 6 demonstrating typical end stage changes of XLRP.
With regard to night blindness, 10 affected males from these families indicated an early onset of difficulty seeing at night (before age 10 years). In fact, these 10 men could not recall ever being able to see at night and often ran into things in the dark when they were children. Two were from RP2 families and eight were RP3. In the remaining four families, affected males did not recall difficulties with night vision until later in their teens. One of these four was RP2 and three were RP3. None of the carrier females complained of difficulty seeing at night.
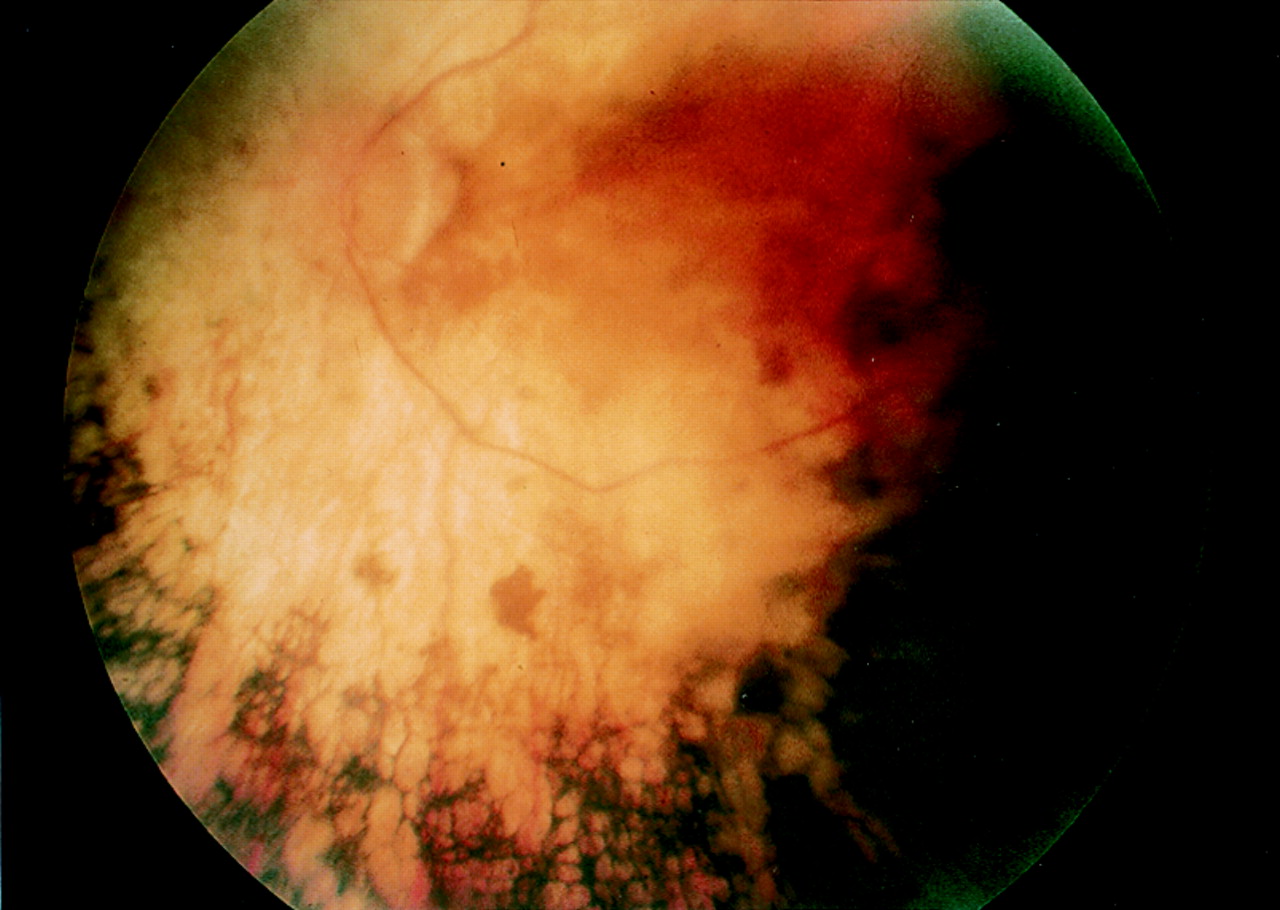
In all 14 families, at least one carrier female was examined and in 10 families, more than one female was examined to give a total of 37 potential female carrier patients. Genetic testing indicated that 33 of those examined were likely to be XLRP carriers. The tapetal reflex was found to be present in carrier females from six of these families. One family with the reflex was RP2 (Fig 2A) and five were RP3 (Fig 2B). Of those carriers where no tapetal reflex was seen, two were RP2 and six were RP3 (Fig 3). When the tapetal reflex was present in one carrier female from a given family, it was generally found in all other carrier females examined from that family. Only in family 14 was a single carrier female found with mild RPE changes and no tapetal reflex, while the other carrier females in the family had the tapetal sheen. This family has been previously described in the literature by Bird.11

(A) Right fundus photograph of carrier female from family 2 with RP2 showing tapetal reflex. (B) Right fundus photograph of carrier female from family 14 with RP3 showing tapetal reflex.
{kind=link}
{kind=link}
{kind=link}
Left fundus photograph of carrier female from family 9 with RP3 showing mild retinal pigment epithelial changes.
Because of the nature of this study, we were not able to perform early electrodiagnostic testing on most patients. Two affected males had electroretinography (ERG) performed before 10 years of age in order to confirm the diagnosis of XLRP. ERGs were otherwise not routinely performed unless it was indicated clinically or in an attempt to clarify carrier status before genetic analysis was available. Visual acuity tests and standard colour vision tests were performed together with clinical history, in an attempt to determine which families showed evidence of early cone dysfunction. Hemizygotic males were questioned regarding the ability to read newsprint and discriminate colours earlier in life to establish early macular cone function. We also asked this question of those females examined. We received no positive answers from any of the females questioned but five of the affected males recalled early difficulty with colour vision and near vision. These males described difficulty reading in school and the need to hold books very close to their eyes. None of them had ERG performed, nor did they have formal colour vision tests. None was highly myopic. Two affected males currently have poor central and colour vision but the age of onset was indeterminate and neither of them had early electrodiagnostic testing or formal colour vision testing. Eight males currently retain good central vision as well as adequate colour vision (at least 75% correct on standard Ishihara colour plates). Of these eight males, two had early electrodiagnostic testing indicating good cone function but absent rod function. One of these families was RP2 and one was RP3 (Table 1). All five families without early electrodiagnostic testing but with early loss of central vision and colour vision by history were RP3.
Discussion
Retinitis pigmentosa (RP) is a variably progressive rod degenerative disorder that shows a high degree of genetic heterogeneity.1 3-8 The X linked form of RP (XLRP) is the most severe form of the disease, typically progressing to severe visual loss in affected males by age 40 years.1 2 8 15 16
Two gene loci have been recognised in XLRP and classified as RP2 and RP3, and a third locus, RP15 has been proposed.8 10 Even within these genetic classifications, there is significant phenotypic variability. As the clustering of XLRP loci on prox Xp often precludes their genetic discrimination, there are families where phenotypic differences would be helpful in classifying the family as RP2 or RP3. Recently, a gene (RPGR) was found to be mutated in seven patients with XLRP8; one of the patients is represented in our review (family 9). A mutation in the RPGR gene has also recently been detected in another family described in this article (family 12); however, no further mutations have been identified in any of our other RP3 families to date. The RPGR gene has been cloned and will facilitate diagnosis in some XLRP families of the RP3 type, but is most likely to be responsible for very few RP3 cases as there have only been a small proportion of XLRP families with RP3 who have the RPGR mutation.8 Phenotypic variations will still be important in the clinical setting when evaluating families thought to have XLRP who are not found to carry the RPGR gene. Even in the present setting of genetic mapping studies and continued identification of more genes and gene mutations, clinical evaluation and phenotype remain important.
The hemizygotic males in our families had varying degrees of myopia compatible with previous reports of XLRP.2 15-18 We found only one family with a significant history of low myopic refractive error in the carrier females.11 One of our families (family 14) has been previously reported13 and it is the one family in which the affected males are all highly myopic. In their report of nine French XLRP families, Kaplan and co-workers found that the RP2 genotype was associated with early, severe myopia in four of the families described.7 Our 14 families do not support this finding as none of the affected males in our families gave a history of early, severe progressive myopia as did the hemizygotic males described by Kaplan’s group and only one of our families had males with high myopia. The distribution of myopia in our hemizygotic males was seven from RP3 families and one from RP2 families. This variation in distribution could be due to the difference in the proportions of RP3 and RP2 families in our series and does not necessarily signify that myopia is more prevalent in RP3.
We found a fairly even distribution of early versus late onset night blindness among our group of families, with both RP2 families and RP3 families showing some variability in the age of onset of night blindness. Kaplan’s group also reported that the RP3 genotype is associated with late onset night blindness in five of their French XLRP families.7 Our data show a slight tendency towards early onset night blindness in the RP3 families with eight of 10 affected males from these families reporting early onset of night blindness, though again our families are weighted towards the RP3 genotype and this possible association may be purely by chance.
It has been well documented by other authors2 11 14-16that hemizygotic males can have extensive loss of central vision by 40 years of age. Jacobson and co-workers reported an RP2 family with an early cone photoreceptor dysfunction documented by psychophysics and electroretinography.9 Although we were unable to carry out their extensive electrodiagnostic tests of cone function on our families, we obtained subjective data similar to that obtained in earlier reports regarding central visual acuity, reading vision, and colour vision.2 16 Five hemizygotic males reported significant reading problems in childhood, generally noted in school, with progressive reduction in central vision and difficulty discerning colours. All of these males were from RP3 families. Because our families are weighted towards RP3, it is difficult to conclude that early onset of cone dysfunction is seen primarily in RP3. We can conclude that some XLRP hemizygotes have an early onset cone dysfunction, but it can occur in either RP2 or RP3. Souiedet al recently reported nine XLRP families with severe manifestations in carrier females associated with the RP3 genotype. None of our families had significant manifestations in carrier females.13 It is likely that these families described by Souied et al are displaying an X linked dominant pattern rather than the X linked recessive pattern, which is the more common manifestation and is that seen in our families.
In conclusion, our review of the phenotypes in this largest reported series of XLRP families classified as RP2 or RP3 by genotype analysis shows that there is significant interfamilial variability in the XLRP phenotype, though within a given family the phenotype is usually homogeneous. We found no clinical features that can be used to reliably differentiate RP2 from RP3 in XLRP and the possibility of more than one abnormal allele at each locus has not been excluded.
Acknowledgments
Presented in part as a poster at the American Academy of Ophthalmology annual meeting, San Francisco California, USA, November 1994.
The authors have no proprietary interest in any devices utilised in this study.
This work was supported by the Medical Research Council, London, grant G9301094.