Article Text

Abstract
Aim: To correlate the phenotype of X linked congenital stationary night blindness (CSNBX) with genotype.
Methods: 11 CSNB families were diagnosed with the X linked form of the disease by clinical evaluation and mutation detection in either the NYX or CACNA1F gene. Phenotype of the CSNBX patients was defined by clinical examination, psychophysical, and standardised electrophysiological testing.
Results: Comprehensive mutation screening identified NYX gene mutations in eight families and CACNA1F gene mutations in three families. Electrophysiological and psychophysical evidence of a functioning but impaired rod system was present in subjects from each genotype group, although the responses tended to be more severely affected in subjects with NYX gene mutations. Scotopic oscillatory potentials were absent in all subjects with NYX gene mutations while subnormal OFF responses were specific to subjects with CACNA1F gene mutations.
Conclusions:NYX gene mutations were a more frequent cause of CSNBX than CACNA1F gene mutations in the 11 British families studied. As evidence of a functioning rod system was identified in the majority of subjects tested, the clinical phenotypes “complete” and “incomplete” do not correlate with genotype. Instead, electrophysiological indicators of inner retinal function, specifically the characteristics of scotopic oscillatory potentials, 30 Hz flicker and the OFF response, may prove more discriminatory.
- X linked congenital stationary night blindness
- genotype-phenotype
Statistics from Altmetric.com
Congenital stationary night blindness (CSNB) encompasses a group of non-progressive retinal disorders in which there is poor night vision. Autosomal dominant (adCSNB), autosomal recessive (arCSNB), and X linked (CSNBX) forms of CSNB have been described.
CSNBX is clinically and genetically heterogeneous. The characteristic electrophysiological abnormality seen in affected males is the “negative wave” electroretinogram (ERG) described by Schubert and Bornschein, in which the b-wave to a-wave ratio is <1.1,2 This finding indicates that CSNBX is an inner retinal dystrophy. A subclassification of CSNB has been proposed based on the psychophysical and electrophysiological characteristics of affected individuals.3 This classification, described before the standardisation of electrophysiological protocols, clinically distinguished the “complete” form from the “incomplete” form primarily by the absence of detectable scotopic ERG responses in the former.
Two causative genes (CACNA1F and NYX) for CSNBX have now been identified through positional cloning strategies.4–7 Mutations in the gene CACNA1F and, more recently, in the gene NYX have been reported in subjects with CSNBX. Although these studies have linked NYX gene mutations with the “complete” form of CSNBX and CACNA1F mutations with the “incomplete” form, electrophysiological and/or psychophysical evidence for a functioning rod pathway has been reported in subjects with NYX gene mutations.6,7 The presence of a genotype-phenotype association requires further investigation as there are reported observations of both “complete” and “incomplete” forms within the same family.8–10 The aim of this study was to address this issue by performing standardised investigation of phenotype in genotyped subjects.
METHODS
Appropriate informed consent was obtained from patients and relatives for clinical and genetic investigations, conforming to the tenets of the Declaration of Helsinki. Twelve families with the clinical features of CSNB and an X linked pedigree structure were initially identified and examined. Clinical testing was performed before the information regarding genotype was completed. Gene mutations in either NYX or CACNA1F were identified in 11 of the 12 families. Following completion of clinical testing, subjects from each pedigree with an identified gene mutation were assigned to one of two groups for phenotypic comparison. Given the small number of subjects involved and the preponderance of subjects with NYX gene mutations, quantitative statistical methods were not appropriate and descriptive statistics have been used to compare the two groups.
Mutation screening
Genomic DNA was extracted from peripheral blood leucocytes using the Nucleon II kit (Scotlab Ltd, Strathclyde, UK) according to the manufacturer’s instructions. Affected male subject DNA was polymerase chain reaction (PCR) amplified for the 48 exons of CACNA1F using primers and conditions previously described.5 Exon 2 of NYX was analysed as previously described7 and exon 3 primers were redesigned (3aF: gacctttggctgacggttgc and 3aR: gtgcaggtagcgcaggtcg in 15 mM MgCl2; 3bF: acaacctgtccttcatcacgc and 3bR: caggttgagcggcgcagg in 10 mM MgCl2; 3cF: gactgtggcgtcctggagc and 3cR: ggtcacctggctgaggtcc in 10 mM MgCl2; 3dF: atggagggctccggacgtg and 3dR: ttaccacaaacacactcaagcc in 15 mM MgCl2). All exon 3 reactions were annealed at 60°C with 1 μl of DMSO in NH4 Reaction Buffer (Bioline, London, UK). Exon 1 primers were designed (1F: acctttacttctctctcaaacca, 1R: ggcatcactgacaacccagc) and used to amplify a non-coding fragment of 260 bp at 60°C annealing temperature.
PCR products were then purified, cycle sequenced and electrophoresed on an ABI 373A automated sequencer as previously described.11 The possibility of a founder effect was evaluated by amplification of microsatellite markers around the NYX gene in affected males from the families sharing the same mutations, as previously described.12
The phenotype of the CSNBX patients was defined by prospective clinical examination of 20 affected males from the 11 families with identified NYX (eight families) or CACNA1F (three families) gene mutations, and by electrophysiological and preliminary psychophysical testing of at least one affected member of each pedigree.
Psychophysics protocol
Colour vision was tested using Ishihara plates, City University plates, and the Mollon-Reffin “minimalist test.”13 A Humphrey field analyser (HFA) was used for photopic perimetry. Scotopic perimetry, spectral sensitivity assessment, and dark adaptometry were performed on nine affected adults from the 11 pedigrees. The modified HFA apparatus and method used have been described in detail previously.14,15 Threshold responses were measured to red (650 nm) and blue (450 nm) targets for dark adaptometry and scotopic perimetry, and additional stimuli of wavelength 488 nm, 520 nm, 590 nm, and 633 nm for spectral sensitivity assessment. The stimulus size corresponded to the Goldmann size V target. Threshold intensity measurements for dark adaptometry and spectral sensitivity were determined at two preassigned retinal locations (−9, +9 and +9, −9 on the HFA 30–2 grid).
Electrophysiology protocol
ISCEV standard ERGs were performed using gold foil electrodes located in the inferior conjunctival fornix in a similar manner to a previously described protocol.16,17 The 10 μs xenon flash was delivered via a Ganzfeld sphere. The maximum intensity developed by the stimulator was 7 cd s−1 m−2. Stimulus intensity was controlled by means of neutral density filters. The pupils were dilated with cyclopentolate 1% and the eyes dark adapted for 20 minutes. The ERG recording electrodes were then positioned under dim red illumination and rod responses recorded to four stimulus intensities. The inter-stimulus interval was 4 seconds for dimmer stimuli and 20 seconds for the standard flash (SF) stimulus. Scotopic oscillatory potential amplitudes were isolated by passing the ERG response elicited in dark adapted conditions using the standard flash through a 100–300 Hz digital filter. The eyes were then exposed to a rod desensitising white background of 25 cd m−2 for 10 minutes following which cone responses were recorded to a white flash at SF intensity and a 30 Hz flicker. The 30 Hz flicker latency was measured from stimulus onset to the first response trough. ON-OFF responses were elicited using a long flash stimulus. A background illumination of 48 ph cd m−2 was used to saturate the rod system, the stimulus intensity was 398 ph cd m−2 and the flash duration was controlled by an electronic shutter with 90% rise and fall times of 5 ms. The duration of the light period was 200 ms and the inter-stimulus interval was 150 ms. The data acquisition sweep time was 300 ms and 50 responses were averaged for each trial. Cursor measurements of the a- b- and d-waves were made on the offline average of two (or more) trials.
Psychophysical and electrodiagnostic tests were performed in the same eye for each subject, selected by optimal visual acuity measurement.
RESULTS
Genotype analysis
Comprehensive mutation screening of the CACNA1F gene revealed three different mutations in three families (Table 1). A novel nonsense mutation was detected in exon 7 (pedigree X10), a single base deletion in exon 9 (pedigree X11), and a nonsense mutation in exon 24 (pedigree X12), all predicted to cause protein truncation and loss of function.
Mutations identified in CSNBX patients
A total of five different mutations segregating with disease were identified in the NYX gene in eight families (Table 1). A splice site mutation, G→C, was detected in family X03 at position +1 in intron 2. A missense mutation identified in two families (X01 and X07) and a nonsense mutation found in family X08 were predicted to cause protein truncation. Two deletions were identified; an in-frame 15 bp deletion in exon 3a in two families (X04 and X06) and a 335 bp deletion in exon 3c−d in two families (X05 and X09).
Clinical examination
All subjects experienced difficulty with night vision but this had remained stationary with age. The visual acuity had deteriorated in three subjects secondary to concurrent advanced glaucoma, myopic macular degeneration or bilateral cataracts. The mean logMAR score was 0.4 (SD 0.2) units (6/18 equivalent, range 6/9−6/60) in both genotype groups. Ten of the 14 (72%) NYX mutation subjects and five of the six (83%) CACNA1F mutation subjects had horizontal nystagmus. Strabismus was apparent in 12 of the 14 (86%) subjects with NYX mutations (esotropia in eight, exotropia in three) and all six subjects with CACNA1F mutations were esotropic. All 20 subjects were myopic with a mean spherical equivalent of −7.25 (SD 4.5)DS in the NYX mutation group and −8.0 (SD 1.5)DS in the CACNA1F mutation group. Mild iris translucency was present in one subject in the NYX mutation group. Tilting of the optic disc with inferotemporal fundal hypopigmentation was seen in the majority of subjects from both genotype groups.
Psychophysical examination
Fourteen of the 20 subjects had normal colour vision to the three testing methods used. Non-specific colour vision abnormalities were identified in six subjects with NYX mutations and one subject in the CACNA1F mutation group. Of these, four subjects had concurrent ocular pathology such as myopic maculopathy, glaucoma, or cataract. Photopic perimetry identified normal or near normal foveal sensitivities but most subjects demonstrated an exaggerated reduction in sensitivity with eccentricity from the fovea compared to normal, non-myopic subjects. Central sensitivities were reduced in the subject with myopic maculopathy and extensive visual field loss was present in the subject with glaucomatous disc. There was no evidence of visual field constriction or mid-peripheral scotomata.
Scotopic static perimetry, spectral sensitivity, and dark adaptometry were performed on nine of the adult subjects without concurrent ocular pathology from the 11 pedigrees, seven from the NYX mutation group and two from the CACNA1F mutation group. All nine subjects tested had nystagmus, visual acuity of 6/9 to 6/18, and ranged in age from 36 to 54 years. Representative examples of the scotopic perimetry results are shown in Figure 1. Mean threshold sensitivity measurements were calculated by averaging the values measured at retinal locations −9, +9 and +9,−9 on the HFA grid. The range of mean threshold elevation in subjects with NYX mutations was 2.5–4.4 (mean 3.5) log units with the blue and 1.6–3.1 (mean 2.0) log units with the red stimulus. CACNA1F mutation subjects showed a range of mean threshold elevation of 1.3–2.3 (mean 2.0) log units for the blue stimulus and 1.8–1.9 (mean 1.9) log units for the red stimulus. In every subject there was a relative reduction in sensitivity in the superior visual field—that is, the inferior retina— of up to 1 log unit compared to the inferior field. The spectral sensitivity curves of subjects from both genotype groups fitted the CIE (Commission Internationale de l’Eclairage) scotopic sensitivity curve without a shift in peak sensitivity towards longer wavelengths.

Scotopic perimetry (representative examples). Mean threshold elevation to a blue stimulus was 3.5 log units in the NYX group and 2.0 log units in the CACNA1F group, whilst that to a red stimulus was 2.0 and 1.9 log units, respectively.
Representative dark adaptation curves for a control and the six CSNBX subjects with reliable responses are illustrated in Figure 2. The mean absolute threshold for the two retinal test locations in NYX mutation subjects was elevated from the control value by 2.4–4.3 log units for the blue stimulus and 1.9–3.2 log units for the red stimulus. In subjects with CACNA1F mutations the absolute threshold for the blue stimulus was elevated by 0.6–1.5 log units and for the red stimulus by 0.7–1.8 log units. Four of the seven NYX mutation subjects and one of the two CACNA1F mutation subjects tested showed a rod-cone break for the blue stimulus, occurring after approximately 9 minutes of dark adaptation. Rod-cone breaks in the remaining subjects could not be reliably identified. The dark adaptation curve of subject X10 III-3 showed no rod-cone break and is likely to have resulted from inadequate bleaching because of the subject’s cataracts. All subjects, regardless of genotype, showed elevated final thresholds but normal adaptation kinetics.

Dark adaptation on curves: blue stimulus, retinal location −9, 9. (A) Subjects with NYX gene mutations. (B) Subjects with CACNA1F gene mutations.
Electrophysiological testing
Electrophysiological results are reported as abnormal if the value was above or below the 95% upper and lower confidence limit of the control group respectively.
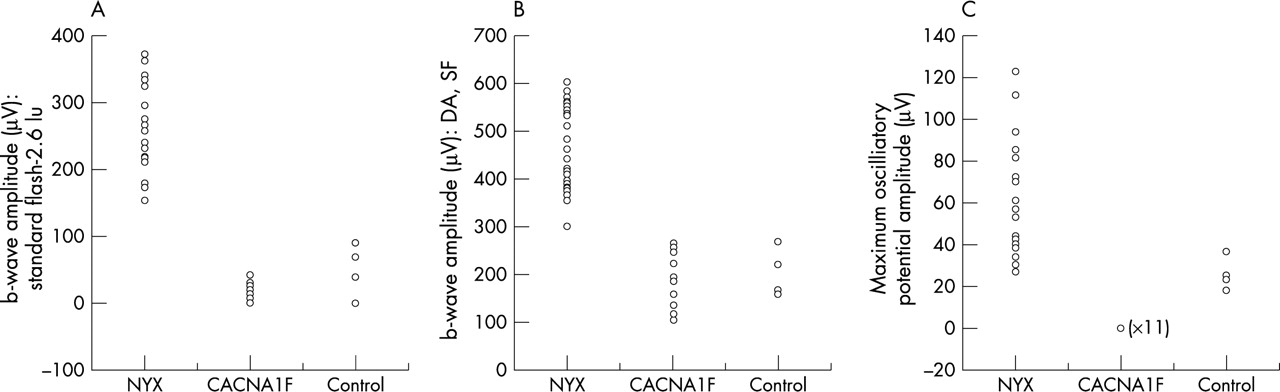
All CSNBX subjects demonstrated a “negative wave” pattern of ERG to a standard flash stimulus (Fig 3B). The b-wave amplitude was subnormal in all subjects with CSNBX, but the mean b-wave amplitude was comparable between genotype groups (Table 2, Fig 4). Two of the 11 NYX mutation subjects had subnormal a-wave amplitudes. The mean a-wave and b-wave latency of the rod mediated ERG was normal in both genotype groups. The waveform in NYX mutation subjects was smooth and devoid of the superimposed wavelets of the oscillatory potentials (OPs) seen in the normal response (Fig 3B). Of the four peaks seen in the normal digitally filtered response (Fig 3C), only two identifiable peaks were seen in subjects with CACNA1F mutations and none of the subjects with NYX gene mutations demonstrated a measurable OP response (Fig 3C). In the absence of four distinct OP peaks in the CACNA1F mutation group, the maximal OP peak amplitude was measured and compared to the maximal OP peak (OP1 or 2) in the control group. The mean maximal OP amplitude was considerably lower in the CACNA1F mutation group than the control group but was unrecordable in all 11 subjects with NYX gene mutations.
Scotopic ERG properties

Comparison of ERG waveforms between genotype groups. (A) A scotopic b-wave response to an SF-2.6 log unit stimulus was recorded in both CSNBX genotype groups but was of lower amplitude in the NYX group. (B) Responses of CSNBX subjects to an SF stimulus in dark adapted conditions showed the typical “negative wave” response. The waveform in the NYX group was smooth and devoid of the wavelets of the oscillatory potentials seen in the normal and CACNA1F mutation group. (C) Scotopic oscillatory potentials (OP) were unrecordable in the NYX group and subnormal in the CACNA1F mutation group. (D) The 30 Hz photopic waveform was saw toothed in the NYX group, and biphasic and subnormal in the CACNA1F response. (E) The ON (b-wave) response was subnormal in all CSNBX subjects. The OFF (d-wave) response was normal in the NYX subjects and borderline subnormal in both CACNA1F subjects tested.

Scatter plots of scotopic ERG responses. (NYX gene mutation group = CSNB1, CACNA1F gene mutation group = CSNB2). (A) Plot of b-wave amplitude to scotopic stimulus (B) Plot of b-wave amplitude to SF stimulus. (C) Plot of maximal OP amplitude.
The a-wave latencies in photopic conditions using the SF stimulus were prolonged in all 11 subjects with NYX gene mutations and three of the four CACNA1F mutation subjects (Table 3). The a-wave amplitude was subnormal in two of the four subjects with CACNA1F mutations. Photopic b-wave amplitudes were also subnormal in two of the four members of this group. Subjects with CACNA1F mutations were extremely photophobic to the 30 Hz flicker stimulus and two subjects from this group were unable to keep their eyes open during recording. Response latency was prolonged in the majority of CSNBX subjects. The response waveform in subjects with NYX gene mutations lacked the OP wavelets on the ascending limb of the response seen in controls and the response from subjects with CACNA1F mutations was double peaked (Fig 3D). The response amplitude was subnormal in both the CACNA1F mutation subjects who tolerated testing.
Photopic ERG properties (SD)
ON-OFF responses were tested in two subjects from each genotype group. A-wave latencies were prolonged in all CSNBX subjects. The d-wave latency was borderline in the subjects with NYX gene mutations but considerably delayed in both subjects with CACNA1F mutations. The a-wave amplitude was subnormal in one of the two subjects with CACNA1F mutations and the b-wave amplitude was subnormal in all CSNBX subjects. The mean d-wave amplitude was considerably reduced in the CACNA1F mutation group compared to the control and NYX mutation subjects, being borderline subnormal in both subjects (Fig 3E, Fig 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scatter plots of photopic responses. (A) Plot of photopic b-wave amplitudes (ON response). (B) Plot of d-wave amplitudes (OFF response).
DISCUSSION
The recent cloning and identification of mutations in the CACNA1F and NYX genes in individuals with CSNBX has confirmed the genetic heterogeneity of this disorder. Clinical heterogeneity may, in part, reflect this genotypic heterogeneity but considerable phenotypic variation exists even in those individuals sharing the same CACNA1F mutation, suggesting that other genetic factors may modify the phenotype.18
We have identified three different mutations of the CACNA1F gene in the 11 CSNBX pedigrees studied. The novel nonsense mutation detected in exon 7 (pedigree X10) is predicted to cause premature protein truncation from domain I of the α1F protein. The single base deletion in exon 9 (pedigree X11) and nonsense mutation detected in exon 24 (pedigree X12), predicted to result in premature protein truncation, have been reported previously (Table 1).4,5 In the remaining eight pedigrees tested in this study, five mutations in the NYX gene were identified. These mutations, including a splice site mutation in intron 2, a missense mutation, and three deletions, are all predicted to result in loss of protein function.6,7 Two novel mutations were detected, a nonsense mutation in pedigree X08 (Gln to stop at residue 299) and a splice site mutation in intron 2 (family X03). Three other mutations detected in the remaining six families in this study have also been detected by other groups (Table 1).
A summary of the phenotypic similarities and differences between the genotype groups is shown in Table 4. The clinical features found in our subjects were similar to those described by Miyake et al, and there were no differences between the genotype groups.3 Specific colour vision abnormalities were not identified and the exaggerated reduction in sensitivity with eccentricity from the fovea demonstrated by photopic perimetry in many of our subjects has been shown to be a common association with high myopia.19
Phenotypic similarities and differences (in bold) between CSNBX genotypes
The results of our psychophysical studies suggest that scotopic detection of a blue stimulus can be mediated by rods in both forms of CSNBX. These results are supported by the findings of Bech-Hansen et al, who also found psychophysical evidence for a functioning but impaired rod pathway in subjects with NYX gene mutations.6 In our study, scotopic rod threshold elevation was a feature common to both genotype groups but tended to be more severe in subjects with NYX gene mutations. Similarly, the mean rod b-wave response was subnormal in both genotype groups but the response was more likely to be undetectable in subjects with NYX gene mutations. Individuals with and without a detectable rod b-wave response were found within the same pedigree. Although this is not the first report of detectable rod b-waves in subjects with NYX gene mutations, rod b-waves are more commonly described as absent in this genotype.7,20 Possible explanations for the high detection rate in our study compared with some of the earlier work includes the use of ISCEV standard stimuli with a Ganzfeld system and modern, sophisticated amplifying and averaging techniques in addition to differences in dark adaptation between laboratories. Although the rod b-wave amplitude did not serve to distinguish the two genotypes in this study, a defining characteristic of the scotopic response was the absence of any measurable oscillatory potentials in subjects with NYX gene mutations. Scholl et al have recently reported the rod a-wave to be subnormal in 10 out of 21 eyes of subjects with NYX gene mutations.20 We have also identified a subnormal a-wave response in two of 11 subjects with this genotype.
Both genotype groups demonstrated cone sensitivity loss on dark adaptometry and scotopic perimetry. A subnormal cone mediated a-wave was detected in two of the four subjects with CACNA1F gene mutations and has previously been reported in subjects with “incomplete” CSNBX.21,22 The subnormal 30 Hz flicker amplitude suggests that the photopic inner retinal responses are comparatively more affected in this genotype group. Subnormal ON responses were seen in all four of the CSNBX patients tested. The subnormal and delayed OFF (d-waves) in subjects with CACNA1F gene mutations suggests an abnormality of the cone OFF retinal pathway that appears to be specific to this genotype and has previously been reported in the “incomplete” form of CSNBX.23–25
CACNA1F and NYX encode two disparate retinal proteins, but loss of function mutations in either gene result in night blindness and similar electrophysiological abnormalities. The night blindness resulting from inactivity of the α1F subunit of the voltage gated L-type calcium channel encoded by the CACNA1F gene many be due to the inadequate stimulation of the ON bipolar cells by glutamate, leaving them relatively depolarised or “light adapted” and reducing the synaptic gain in transmission between photoreceptor and bipolar cell. The functional role of the NYX gene product, nyctalopin is not yet understood; as other proteins of the same protein superfamily are involved in regulation of cell growth, cell adhesion, and axon guidance, nyctalopin may regulate synaptic formation.26,27
Recent investigation of the slow and fast rod ERG pathways in subjects with NYX gene mutations has led to the theory that the absence of nyctalopin primarily prevents conduction through the rod bipolar-AII cell (slow) pathway, causing severe reduction in the rod ON bipolar response (b-wave).20 The presence of a subnormal photopic ON response suggests an additional effect on the cone photoreceptor-ON bipolar pathway but no apparent effect on the cone OFF bipolar response. Mutations in the CACNA1F gene also appear to affect the rod photoreceptor-AII pathway and the cone-ON bipolar pathway but additionally interfere with the cone-OFF cone bipolar pathway as illustrated by the subnormal OFF response. The subnormal 30 Hz flicker response and subnormal a-wave amplitudes evident in individuals with this genotype imply that multiple anomalies are present in the inner retinal circuitry.
This is the first study in which standardised examination has enabled phenotypic comparisons between genotyped subject groups. “Complete” CSNBX has been characterised primarily by the absence of a scotopic response.3 In this study, nine of the 11 subjects with NYX gene mutations tested demonstrated evidence of a functioning but impaired rod pathway and would therefore be clinically labelled as having “incomplete” CSNBX. These results show that the severity in the reduction of scotopic function alone does not appear to be a reliable indicator of genotype: “complete” and incomplete” phenotypes do not correlate with genotype. More useful clinical indicators of genotype may include absent scotopic oscillatory potentials in subjects with NYX gene mutations, and the subnormal OFF cone bipolar response, which appears specific to subjects with CACNA1F gene mutations.
ELECTRONIC DATABASE INFORMATION
Accession numbers and URLs for data in this article are as follows: NYX gene sequence, AJ278865, and CACNA1F gene sequence, AF067227, AJ224874, and AJ002616 all from Genbank at www.ncbi.nlm.nih.gov/
Acknowledgments
The authors thank all participating families for their cooperation and thank Professor John Mollon and Mr Vy Luong for their contributions to the project. This research was funded by the Wellcome Trust and The Guide Dogs for the Blind Association.