Article Text

Abstract
Human congenital cataract has a diverse aetiology. In the proportion of cases where the cause is genetic, the disease shows wide phenotypic and genetic heterogeneity. Over the past few years, much research has been devoted to mapping the genes that underlie the disorder. This has been helped by the extensive array of naturally occurring and genetically engineered mouse cataract models and the abundance of human candidate genes. Most progress to date has been in the identification of genetic mutations causing autosomal dominant congenital cataract where eight genes have been implicated in cataractogenesis. Overall there is good correlation between the genetic mutations so far identified and the resulting lens phenotype but it is clear that mutations at more that one locus may give rise to similar forms of cataract.
The identification of genes causing inherited forms of cataract will improve our understanding of the mechanisms underlying cataractogenesis in childhood and provide further insights into normal lens development and physiology. Perhaps more importantly, it is likely that some of the genes causing early onset cataract will be implicated in age related cataract which remains the commonest cause of blindness in the world.
- cataract
- congenital
- genetics
- phenotype
Statistics from Altmetric.com
Cataract is the term used to describe opacification of the crystalline lens of the eye. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. In general, the more posteriorly located and dense an opacity, the greater the impact on visual function.1
Cataract is the commonest treatable cause of visual disability in childhood,2 3 with an incidence of 1-6 per 10 000 live births. There are many different causes including intrauterine infections, metabolic disorders, and chromosomal abnormalities.3 Cataract may also be inherited either as an isolated ocular abnormality or as part of a syndrome. The syndromic forms of cataract, which have recently been reviewed,4will not be covered in this paper. In non-consanguineous populations, the majority of inherited non-syndromic cataract shows autosomal dominant (AD) inheritance, but X linked and autosomal recessive forms are also seen.4
Inherited non-syndromic cataract phenotypes
Classification of human inherited cataract is difficult because of the wide variation in morphologies observed.5 The lens develops by the formation of an embryonic nucleus during morphogenesis, around which lens fibres are deposited throughout life, initially forming the fetal nuclear region and thereafter the cortex (fig 1). Animal models suggest that the genes so far implicated in cataractogenesis are expressed in a time ordered, sequential fashion.6 Categorisation, therefore, more weighted towards the location of opacification rather than appearance, will accommodate these developmental considerations and best reflect the underlying genotype. Such a system is also clinically convenient.
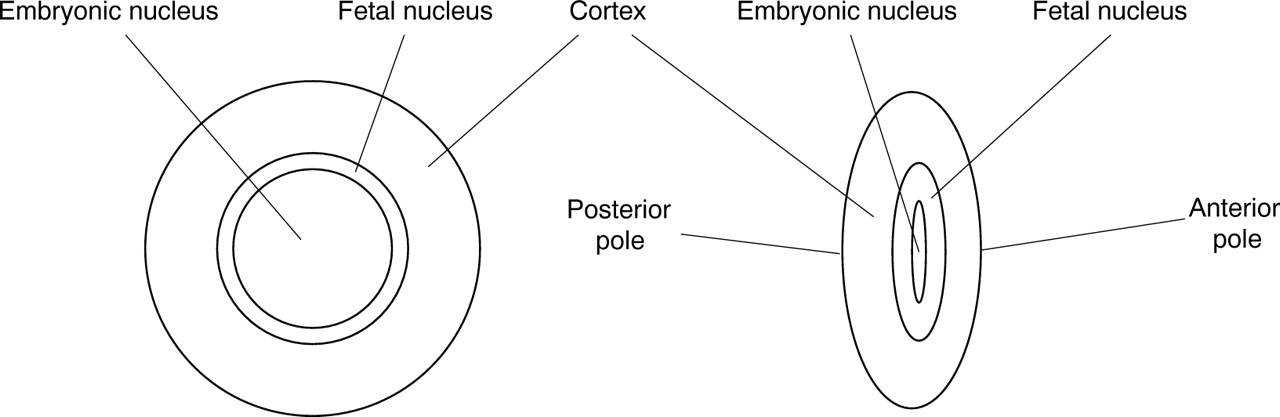
The human crystalline lens.
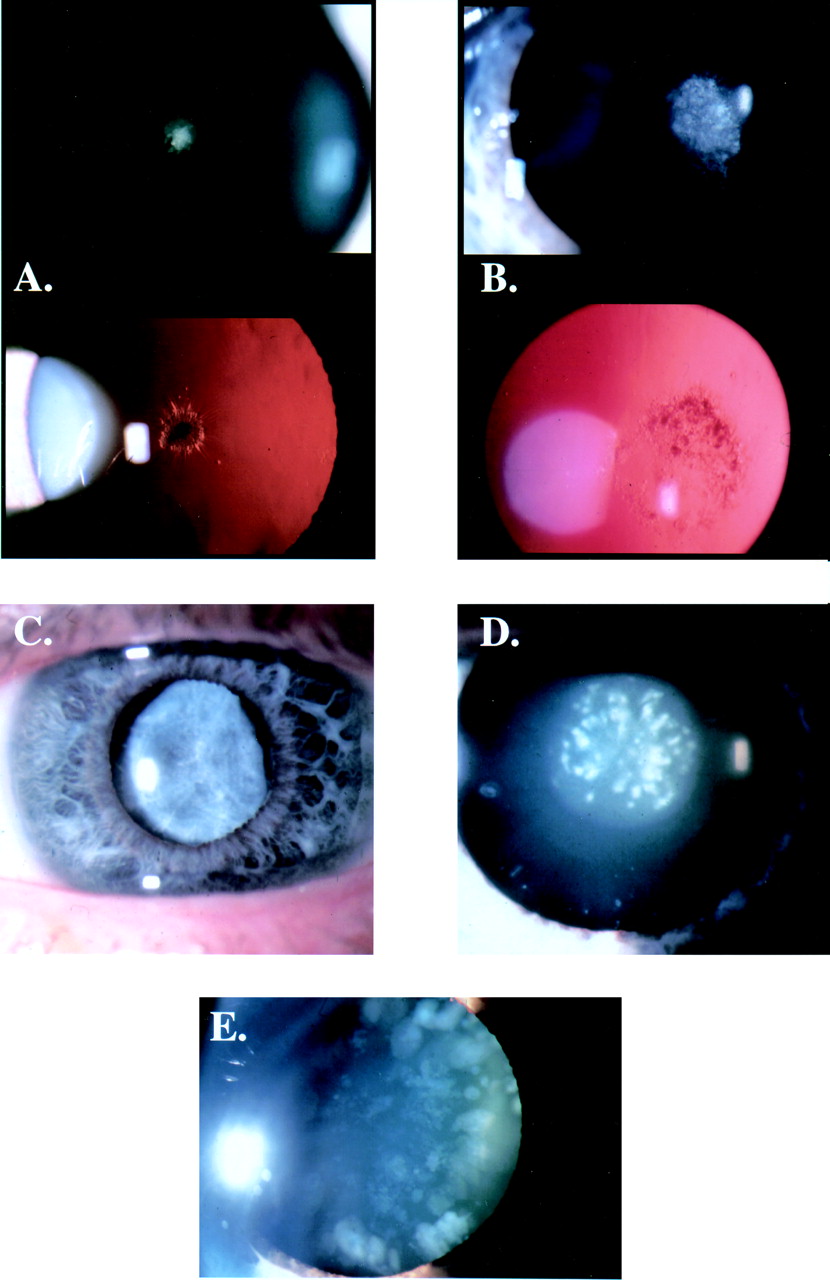
Cataract affecting the nucleus (fig 2C) is common and suggests an abnormality of gene expression in early development. Opacities may be confluent or discrete. Affected subjects show bilateral symmetrical involvement with variable expressivity. An exception is the pulverulent cataract where the type and distribution of the nuclear opacities can vary not only between family members but also between eyes of the same patient.5
{kind=link}
{kind=link}
Examples of inherited cataract phenotypes. (A) Discrete non-progressive central anterior polar cataract (24 year old female). (B) Non-progressive posterior polar cataract (9 year old male). (C) Nuclear opacification (15 year old male). (D) Fine, dust-like (pulverised) opacities in lens with pulverulent cataract (32 year old male). (E) Discrete progressive blue-white pinhead and wedge shaped opacities typical of the blue dot or cerulean cataract (45 year old female).
Pulverulent cataract (fig 2D) derives its name from the dust-like “pulverised” appearance of the opacities which can be found in any part of the lens. The first detailed description of an affected family was published by Nettleship and Ogilvie7 in 1906. In this, the Coppock family, the cataract was confined to the embryonic nucleus and has been termed central pulverulent,8 in all probability the phenotype previously described as Doyne's discoid cataract.9 10 The Coppock family has not been the subject of a published linkage study, unlike the genealogically unrelated pedigree with cataract described as Coppock-like,9 11 12which has been linked to the crystallin gene cluster region on 2q. It is of note that the Coppock phenotype and the cataract investigated by Renwick and Lawler in the “Ev family from southern England”13 14 have become synonymous. However, the latter involves the larger fetal nucleus with opacification increasing in density towards the periphery and is therefore identical to the family with zonular pulverulent cataract described by Poos.15 16
Many other families with pulverulent cataract have now been described.7 17-19 It is clear that significant intra- and interfamilial variation, both in the distribution of the cataract and the degree of opacification, distinguish this phenotype from all others.
The concentric deposition of secondary lens fibres that occurs during growth of the normal lens results in the formation of lamellae. Opacities confined to a specific lamella therefore reflect a short period of developmental disturbance (usually during the fetal period) resulting in symmetrical bilateral lens opacification. Lamellar cataracts have also been called zonular, perinuclear, polymorphic,20 or Marner's cataract.21 The degree of opacification is variable and visual acuity may be well preserved or reduced enough to require surgical intervention.22 Commonly, cataract occurs at the anterior and posterior Y sutures. In some cases, cortical opacities or “riders” are associated with lamellar cataract.
Cataract limited to the cortex is rare and differs from lamellar cataract since opacification is restricted to a sector of outer cortical, often superior, lens fibres, adjacent to the lens capsule. The nucleus is unaffected. The pathogenesis is unknown but its distribution and subsequent progression suggest an abnormality of the later stages of lens development.
The presence of families with cataract limited to either the anterior or posterior pole of the lens is less amenable to explanation in terms of lens development. Anterior polar cataracts (fig 2A) are bilateral, usually symmetrical, well circumscribed lens opacities that are rarely progressive and can be inherited as dominant, recessive, or X linked traits.23 24 Larger opacities often have a pyramidal shape, the apex of which may extend into the anterior chamber.5 25 26 Associations with microphthalmia27 and astigmatism28implicate a gene involved in anterior segment development. Visual function is usually well preserved.29 Families with posterior polar cataracts (fig 2B) are reasonably common. Affected subjects have bilateral, symmetrical lens opacities which are usually inherited as a dominant trait. Since opacification is close to the optically crucial, nodal point of the eye, vision is commonly reduced.30 In some families, progressive accumulation of further posterior cortical opacities can lead to total cataract formation.10 14 25 31
The blue dot (cerulean) cataract (fig 2E), first described by Vogt,32 is not truly congenital, but develops in childhood and progresses through early life.33 The discrete, pinhead shaped, blue-white opacities are distributed throughout the lens becoming more numerous in the cortex where they may form large cuneiform (wedge-like) shapes in the mid-periphery. Within a pedigree, this phenotype is consistent in its distribution but variable in its severity. Acuity is usually well preserved; cataract extraction is rarely necessary before adult life and is usually associated with a good outcome.8 34
A peculiar and rare form of cataract, “coralliform” or “aceuliform”, originally described by Nettleship,35 is characterised by finger-like protuberances extending from the nucleus that resemble sea coral.10 36 The visual impact is variable but cataract extraction is usually required in the early years of life.
“Total” cataract, that is, lens opacity apparently affecting both nuclear and cortical regions, has been reported in families both with autosomal dominant37 as well as X linked recessive congenital cataract.38 It has also been reported as the end result of the progression of the phenotypes outlined above. Other uncommon phenotypes have been described in isolated cases, but not documented in families.
The molecular genetics of inherited cataract
In 1963, Renwick and Lawler13 described in their seminal publication the cosegregation of inherited cataract with the Duffy blood group locus (Online Mendelian Inheritance in Man OMIM reference number 110700). This became the first autosomal disease to be genetically linked in man when, in 1968, the Duffy locus was assigned to chromosome 1.39 Subsequent development of advanced molecular biological techniques has facilitated the identification of 20 further independent cataract loci (table 1) including 10 mutations (table 2). In most cases a candidate gene approach has been used once linkage has been established (table 3). There are, however, several practical considerations when mapping human cataract genes. A significant proportion of cataract mutations appear de novo often making family size small. While penetrance in all phenotypes is high, expressivity, age of onset, and rate of progression are variable, making careful ophthalmic evaluation critical. In addition, surgical modification of the disease can make it difficult to describe the phenotype accurately.
Mapped loci for human congenital non-syndromic cataract with no candidate gene. Evidence for linkage to each locus is based upon the publication of single family data
Identified human congenital cataract mutations
Candidate genes for human congenital cataract
GENES IMPLICATED IN CATARACTOGENESIS
Recently, the Duffy blood group locus has been refined to 1q22-2340 and a mutation identified in the gene coding for connexin50 in a family with autosomal dominant pulverulent cataract.41 The gene was considered an ideal candidate since it is abundantly expressed in the human lens, its protein product forming an integral part of the extensive gap junction network of lens fibre membranes. Connexins are a diverse family of molecules that associate into heterogeneous oligomeric transmembrane structures with a central voltage gated ion channel, known as connexons. Connexons bridge the extracellular space allowing the passage of small molecules between adjacent cells.42 A feature of the mature lens cell is its metabolic inactivity. It is likely therefore that the connexin50 mutation which occurs in the highly conserved second transmembrane domain results in altered function with subsequent disruption of cell homeostasis observed as a loss of clarity. Further evidence is provided by the identification of mutations in another gap junction protein, connexin46 on 13q, in two families with pulverulent cataract.43 Mutations of other connexin genes have also been implicated in other inherited disorders, namely Charcot-Marie-Tooth disease, inherited deafness, and congenital cardiac disease.44-46
α-, β-, and γ-crystallins constitute the main cytoplasmic proteins of the human lens. By forming tight packing stable oligomers that interact with the surrounding cytoskeleton (characterised by the presence of a unique beaded filament structure), lens fibre transparency is maintained.47 β- and γ-crystallin genes have also been shown in many species to encode ubiquitous enzymes. α-crystallin has been shown to be a member of the heat shock protein family. This dual use of a distinct protein encoded by a single gene, termed gene sharing, is probably common in the lens and other systems.48 These are clearly strong candidate genes and to date three cataract causing mutations have been identified.
The γ-crystallin gene cluster (2q33-q35) consists of genes γA, B, C, D, E, F, and a gene fragment γG.49 Only γC and γD encode abundant proteins while γE and γF are pseudogenes by virtue of in frame stop codons (the γF lacks a promoter as well). The mutation underlying the (pulverulent) Coppock-like cataract was thought to result in the activation of the γE pseudogene whose product is an N-terminal protein fragment, the deposition of which was likely to cause the cataract.49 Re-evaluation of the original data has recently questioned this view and it is now considered likely that the cataract arises from a missense mutation in a highly conserved segment of exon 2 of the γC-crystallin gene.50
Another missense mutation in the γD-crystallin gene that results in the substitution of arginine for cysteine at codon 14 has been suggested to result in a progressive nuclear cataract.51Protein modelling predicts that the substitution results in subtle changes in the surface properties of the crystallin, consistent with the mild but progressive nature of the phenotype observed.
Crystallin βB2 is the only member of the β-crystallin gene cluster on 22q to be highly transcribed in the lens. Missense mutations in this gene are now known to result in the development of blue dot (cerulean)52 and the Coppock-like cataract.53
Mutations in α-crystallin are now known to be cataractogenic. A splice site mutation54 is thought to be responsible for the sutural (lamellar) opacities observed in a family mapped to 17q55 and a missense mutation in the crystallin αA gene has been identified56 in a family with zonular nuclear cataract.
The report of a mutation within the homeobox gene,PITX3, is the first to implicate a developmental regulator gene in the pathogenesis of congenital cataract. The G to A transition identified results in the substitution of serine for asparagine at codon 13 which, although not within the crucial homeodomain of the protein, is predicted to affect either DNA binding or inhibit protein-protein complex formation. Interestingly, mutations in PITX3 have also been shown to result in anterior segment mesodermal dysgenesis (ASMD, OMIM 107250) in which cataract is encountered in combination with other complex anterior segment abnormalities.37
GENETICALLY MAPPED CATARACT LOCI
It is of interest that many cataract families have been mapped to loci for which there is no known candidate gene (table 1). Two families with autosomal dominant cataract have shown linkage to the 1p36 locus, the first with a posterior polar phenotype25 and the other a large Danish family with progressive zonular and nuclear opacities (probably pulverulent).57 In the latter family, a mutation was sought in τ-crystallin, which is not expressed in the human lens but lies within this locus. Perhaps not surprisingly, no mutation was identified. Another Danish family, first reported by Marner, with primarily lamellar cataract, shows strong linkage to the haptoglobin locus on 16q22.1.
Another three families with dominantly inherited cataracts have been mapped to chromosome 17. The first, with anterior polar cataract, shows linkage to 17p13,26 the second, with lamellar opacities, maps to 17q11-q12,55 distinct from the third family with the blue dot (cerulean) phenotype, mapped to 17q24.58
Anterior polar cataract has also been reported in association with an apparently balanced chromosomal translocation t(2;14) (p25;q24).24 Following the recognition of a female with multiple abnormalities, including congenital cataract in association with a terminal deletion of chromosome 14, it has been argued that a cataract locus must therefore reside in the region 14q24.59
The recognition of another family with a reciprocal translocation has identified a further cataract locus on 16p.60 In this family, a balanced translocation t(2;16)(p22.3;p13.3) was observed in four subjects; three had partial trisomy 2p derived from this translocation and two had a normal karyotype. All patients with translocations had cataracts and those with the normal karyotype had not, suggesting the cataract causing gene lay in the region 16p13.3. Autosomal recessive forms of inherited cataract have been reported in several genealogically distinct populations and seem particularly prevalent in the Japanese. Linkage to the I blood group has been suggested.61
The existence of X linked non-syndromic congenital cataract remains contentious. A number of pedigrees have been reported, though in many other modes of inheritance appear more likely. It has been suggested, however, that X linked cataract is either synonymous or closely related to the Nance-Horan syndrome, mapped to Xp. Furthermore, the recognition of chromosomal deletions of varying size in this region and the resulting phenotypes observed suggest that a cataract locus may reside within Xp22.3-21.1.38
Significantly, exclusion data on other families with autosomal dominant cataract have been reported, strongly supporting the supposition that further genetic loci remain to be identified.5
Mouse models for human cataract
Opacification of the lens is relatively easily detected in mice and this has in part led to the recognition of a number of spontaneously occurring strains with heritable cataract traits. The use of teratogenic agents has also generated a number of mouse cataract models.
Mouse models have made considerable contributions to the field of cataract research. Firstly, they have confirmed the importance of several genes and proteins in the maintenance of lens clarity.62 Secondly, the extensive synteny between the mouse and human genomes has facilitated the identification of novel candidate genes for human cataract formation.63 Thirdly, it is now clear that the lens plays an essential role in guiding normal eye development and the identification of certain mutations has enabled this process to be dissected.64
Table 4 shows those mouse mutants for which there is a known human corollary. It is of note that the phenotypic appearances between the two species often differ. Possible explanations include difficulties in classification, differences in physiology, and alternative effects of different mutations within the same gene and the effects of different modifier genes.
Mouse cataract models for which a human homologue is known
Several other cataract causing mutations have been identified in mice65 and it will be interesting if human homologues are soon identified. The exciting prospect is that mouse cataract models will provide increasingly sophisticated experimental strategies for the study of the human disease.
Genotype-phenotype correlations
The presence of several clearly distinguishable human cataract phenotypes and a number of probable subtypes within each category parallel well the complex underlying genotype shown by human linkage studies. Furthermore, evidence that each phenotype maps to more than one locus suggests mutations in different genes may give rise to similar phenotypes.
In contrast, only γC- and βB2-crystallin genes have to date been implicated in more than one phenotype. It is possible, however, that allelic heterogeneity will be shown to be more prevalent as different mutations within the same gene may affect the regulatory ability of the protein product or its ability to bind with other lens proteins. An example of this might be α-crystallin which is known to have both structural as well as chaperone-like functions. It remains to be seen whether the Volkmann and posterior polar cataract loci identified on 1p36 are indeed allelic.
Lens development and growth throughout life results from the temporal and sequential expression of a number of genes. There is some correlation between what is known about the distribution of proteins in the lens and the position of opacities seen in cataract. An example is the blue dot (cerulean) cataract resulting from a mutation in β-crystallin known to be found in the cortical region of the lens. Much remains to be elucidated in lens biology but the identification of further underlying genetic mutations in patients with cataract will be beneficial.
Genetic counselling
Genetic counselling in congenital cataract is usually straightforward when the abnormality is confined to the lens and there is a positive family history. Most families show autosomal dominant inheritance and the status of at risk subjects can readily be assigned by careful slit lamp examination after pupillary dilatation. Variability in disease expression is common and asymptomatic subjects should not be assumed to be unaffected. X linked and recessive forms of inherited cataract are rare and may be recognised when there is an appropriate family history.
Genetic counselling in isolated cases is more problematical. Most unilateral cataract is non-genetic but patients with bilateral cataract in whom there is no family history should undergo further investigation to elucidate the cause.30 Firstly, both parents and any sibs should undergo dilated slit lamp examination to exclude mild congenital opacities; the presence of such opacities will confirm the familial nature of the cataract and allow accurate counselling of recurrence risks. If other family members are normal, the child should be reviewed by a dysmorphologist or paediatrician to rule out any other clinical features that may suggest a multisystem disorder associated with cataract. Routine investigations include plasma urea and electrolytes, urinary amino acids (to exclude Lowe's syndrome in male infants), urinary reducing sugars (to exclude galactosaemia), and a screen for congenital infection, particularly rubella.30Other investigations may be required depending on other clinical findings. In the absence of a family history and where investigations prove normal, the risk of recurrence in subsequent pregnancies is extremely small.
When counselling adults with congenital cataract about the risk to their offspring, it is again important to review other relatives and where possible examine clinical records to exclude any syndromic forms of cataract or non-genetic aetiology. In adults without a family history, the risk of having an affected child is very small if the cataract is unilateral. The risk is higher in bilateral cases as some may represent new autosomal dominant mutations; the precise risk is difficult to quantify. Many of the adults seeking advice will have had multiple operations in childhood and still have severe visual impairment; they may have reservations about putting their own child through a similar experience. However, improvements in cataract surgery and optical management have resulted in greatly improved visual outcome and multiple operations are rarely necessary.66 This improved prognosis should be discussed and it is important that the newborn child is examined by an ophthalmologist in the first few weeks of life to exclude cataract as the long term prognosis in infants that require early surgery is improved if surgery is performed promptly.
Note added in proof
Recently Berry et al 78have reported that missense mutations in the gene encoding the major intrinsic protein of the lens (MIP) underlie an autosomal dominant form of polymorphic and lamellar cataract in man.
Acknowledgments
The authors would like to acknowledge Wellcome Trust for their support of our research (grant 053416) and Mr Philip Ball, Senior Medical Illustrator, Addenbrooke's Hospital, Cambridge for his help in the preparation of the clinical photographs.