Article Text

Abstract
Background Variation in the ABCA4 gene is causal for, or associated with, a wide range of phenotypes from early onset Mendelian retinal dystrophies to late-onset complex disorders such as age-related macular degeneration (AMD). Despite substantial progress in determining the causal genetic variation, even complete sequencing of the entire open reading frame and splice sites of ABCA4 identifies biallelic mutations in only 60%–70% of cases; 20%–25% remain with one mutation and no mutations are found in 10%–15% of cases with clinically confirmed ABCA4 disease. This study was designed to identify missing causal variants specifically in monoallelic cases of ABCA4 disease.
Methods Direct sequencing and analysis were performed in a large familial ABCA4 disease cohort of predominately European descent (n=643). Patient phenotypes were assessed from clinical and retinal imaging data.
Results We determined that a hypomorphic ABCA4 variant c.5603A>T (p.Asn1868Ile), previously considered benign due to high minor allele frequency (MAF) (~7%) in the general population, accounts for 10% of the disease, >50% of the missing causal alleles in monoallelic cases, ~80% of late-onset cases and distinguishes ABCA4 disease from AMD. It results in a distinct clinical phenotype characterised by late-onset of symptoms (4th decade) and foveal sparing (85%). Intragenic modifying effects involving this variant and another, c.2588G>C (p.Gly863Ala) allele, were also identified.
Conclusions These findings substantiate the causality of frequent missense variants and their phenotypic outcomes as a significant contribution to ABCA4 disease, particularly the late-onset phenotype, and its clinical variation. They also suggest a significant revision of diagnostic screening and assessment of ABCA4 variation in aetiology of retinal diseases.
- Stargardt disease
- ABCA4
- Age-related macular degeneration
- hypomorphic variant
- foveal sparing
Statistics from Altmetric.com
Introduction
Mutations in the ABCA4 gene (STGD1, MIM #248 200)1 are the most frequent cause of Mendelian inherited recessive retinal dystrophies with phenotypes ranging from late-onset mild cases of central atrophy to very early onset, retinitis pigmentosa (RP)-like, panretinal degeneration.2–5 Similar to phenotypic variability, the disease-associated genetic variation in ABCA4 is also extensive.6 Currently, >1000 definitely or possibly disease-causing variants have been determined in the coding sequences and splice sites of the ABCA4 gene (JZ and RA, unpublished data). While the understanding of genetic variation in ABCA4 has substantially improved since the discovery of the gene in 1997, diagnostic screening in patients diagnosed with ABCA4 disease (often collectively referred to as Stargardt's disease) is successful in only 60%–70% of cases where both disease-causing alleles are identified.7 8 The remaining cases include patients with one mutation (20%–25%) and no mutation (10%–15%).7 The population frequency of ABCA4 alleles is high—1:20 people carry a potentially pathogenic ABCA4 allele.7 9 Therefore, recent efforts have been directed towards two goals; first, to find the missing ABCA4 disease-causing alleles in non-coding sequences8 and, second, to perform extensive whole exome sequencing (WES) in patients with 0 ABCA4 alleles (and ABCA4-like phenotypes) to differentiate non-ABCA4 phenocopies.10–13 Substantial progress has been made with respect to both objectives: several deep intronic ABCA4 variants have been proven to affect splicing8 14 and preliminary data (not shown) indicate that such is also true of rare variants affecting ABCA4 expression. We have also shown that large CNVs, which elude detection by sequencing, are exceptionally rare in the ABCA4 locus and, as a result, account for only a small fraction of ‘missing’ alleles.15 WES of patients with 0 ABCA4 mutations has uncovered causal genes in ~70% of cases with ABCA4-like phenotypes, including known retinal disease genes (CRB1, CRX, etc)11 13 and new genes (RAB28, RDH11).10 12 However, despite all these advances, the second causative mutation in most patients with 1 and 0 ABCA4 alleles remains unidentified following complete sequencing.8 This study demonstrates that some known, frequent ABCA4 variants, which have been considered benign due to high minor allele frequency (MAF) in the general population, are in fact hypomorphic alleles, which result in disease expression under certain conditions.
Materials and methods
Patient cohort
All study subjects were consented before participating in the study under the protocols #AAAI9906 approved by the Institutional Review Board at Columbia University and #20130770 by the Western Institutional Review Board at the Chicago Lighthouse. The study adhered to tenets established in the Declaration of Helsinki. Complete ophthalmic examinations were provided by a retinal specialist (SHT and GAF), including slit-lamp and dilated fundus examinations. Clinical assessments (WL and FTC) were made from clinical examination notes, retinal imaging data and research questionnaires. Spectral domain-optical coherence tomography (OCT) scans and corresponding infrared reflectance fundus images were acquired using a Spectralis HRA+OCT (or HRA+OCT) (Heidelberg Engineering, Heidelberg, Germany). Fundus autofluorescence (AF) images were obtained using a confocal scanning-laser ophthalmoscope (Heidelberg Retina Angiograph 2, Heidelberg Engineering, Dossenheim, Germany). Fundus autofluorescence (AF) images were acquired by illuminating the fundus with an argon laser source (488 nm excitation) and viewing the resultant fluorescence through a band pass filter with a short wavelength cut-off at 495 nm. Colour fundus photos were obtained with a FF 450plus Fundus Camera (Carl Zeiss Meditec AG, Jena, Germany) and CR-1 Mark II Fundus Camera (Canon, Tokyo, Japan).
Sequence analysis
The ABCA4 gene and locus sequencing were performed as previously described, using the Illumina TruSeq Custom Amplicon protocol (Illumina, San Diego, California, USA), followed by sequencing on Illumina MiSeq platform, or the RainDance microdroplet-PCR target enrichment (RainDance Technologies, Billerica, Massachusetts, USA), with subsequent sequencing on Roche 454 platform.8 15 The next-generation sequencing reads were analysed and compared with the ABCA4 sequence in the reference genome GRCh37/hg19, using the variant discovery software NextGENe (SoftGenetics, State College, Pennsylvania, USA). All detected possibly disease-associated variants and their segregation with the disease in available family members were analysed and confirmed by Sanger sequencing. Nucleotide positions and protein translation correspond to CCDS747.1 and NP_000341.2, respectively. Nucleotide numbering uses the A of the ATG translation initiation start site as nucleotide 1. The possible effect of all ABCA4 variants was assessed as described in detail previously.8 15 The variant prediction algorithms were accessed via Alamut Visual version 2.7 (Interactive Biosoftware, Rouen, France). The allele frequencies of all variants were compared with the gnomAD database,16 and The 1000 Genomes Project database.17 All ABCA4 variants reported in this manuscript were submitted to Leiden Open Variation Database V.3.0 (www.lovd.nl/3.0/home).
Molecular modelling
Amino acid sequence P78363 (ABCA4_HUMAN) was obtained from the Uniprot database (http://www.uniprot.org). Two transmembrane and nucleotide-binding motifs of the human ABCA4 structure were built by homology modelling similar to that of previously described for the RS1 protein.18 19 Briefly, the homology model of structure of transmembrane and nucleotide-binding domains of ABCA4 was built using the automatic segment matching and self-consistent ensemble optimisation by the Gene Mine Look program, V.3.5.2.18 Bacterial lipid ‘flippase’ MsbA was chosen from the Crystallographic Database as a structural template (PDB: 3b60). Hydrogen atoms were added to the structure and the predicted ABCA4 structure was 2 ns equilibrated in water using molecular dynamics (MD). MD simulations were performed using the Impact module of the Maestro program package (V.8.0.308, Schrodinger, New York, New York, USA). The quality of the structure was accessed with the PROCHECK program. Protein unfolding propensities for mutant variants p.Asn1868Ile and p.Gly863Ala were evaluated using the unfolding mutation screen as previously described in detail.20 21
Results
Analysis of genetic data
The study cohort consisted of 643 individuals of (mostly Eastern) European descent. Of these, 2 ABCA4 mutations were identified in 437 cases (68%), 1 mutation in 117 cases (18%) and 0 mutations in 89 cases (14%) (see online supplementary table 1), leaving ~23% of disease-associated alleles in 32% of patients yet to be identified. Almost all patients with no ABCA4 mutations and ~50% of patients with 1 mutation have been screened by whole exome sequencing to determine if variants in other genes were causal in these cases. All cases, where disease-associated variants in other genes were detected, were excluded from this cohort.
The ABCA4 gene harbours many common missense variants with MAFs >5% in the general population. The c.5603A>T (p.Asn1868Ile) allele was present in patients with ABCA4 disease three to four times more frequently than expected (~20% vs 6.6%), an observation which had also been previously reported.22 23 The allele frequency of the p.Asn1868Ile variant is ~6.6% in Europe, 2% in Latinos and South Asians, and 1% in Africans (gnomAD). This allele was also observed to be in cis with several more frequent ABCA4 disease-causing alleles, notably p.5461–10T>C (p.?) and c.2588G>C (p.Gly863Ala) (table 1 and see online supplementary table 1). Therefore, we assumed that the more frequent occurrence of p.Asn1868Ile in patients with ABCA4 disease is due to linkage disequilibrium with other truly pathogenic alleles. However, when complex alleles with known pathogenic mutations were excluded from these calculations, the allele frequency in patients remained significantly higher.
Genotypes of cases with single ABCA4 pathogenic variants and the c.5603A>T (p.Asn1868Ile) status
In 643 screened patients, p.Asn1868Ile was detected in 176 cases (197 alleles); 21 cases were homozygous and 155 cases heterozygous. When dividing the cohort into three groups based on the number of detected ABCA4 pathogenic alleles as described above, the fractions of the p.Asn1868Ile alleles were 11.6% (101/874) in 2 mutation cases, 33% (77/234) in 1 mutation cases and 10.6% (19/178) in 0 mutation cases. Therefore, the allele frequency of p.Asn1868Ile was two times higher as compared with the matched general population in cases with 2 and 0 mutations and 5X higher in cases with 1 mutation (33% vs 6%; p<0.0001). More frequent disease-causing alleles which always carried p.Asn1868Ile in cis were c.5461–10T>C (p.?), c.4469G>A (p.Cys1490Tyr), c.4594G>A (p.Asp1532Asn) and c.2588G>C (p.Gly863Ala) (see online supplementary table 1). Other, rarer disease-causing variants which were found allelic with p.Asn1868Ile after determining the phase included c.319C>T (p.Arg107*), c.1253T>C (p.Phe418Ser), c.2552G>A (p.Gly851Asp), c.5114G>A (p.Arg1705Gln), c.5318C>T (p.Ala1773Val) and c.5572T>A (p.Tyr1858Asn).
After accounting for known disease-associated alleles that harbour the p.Asn1868Ile variant in cis, we determined that over half of the cases with 1 mutation (67/117, 57.3%) carried the variant in trans (table 1). We then assessed all 67 cases of whom 57 were heterozygous for the p.Asn1868Ile variant and 10 were homozygous. The homozygous fraction included cases with p.Asp1868Ile as a complex allele, mostly due to the c.5461–10T>C variant (n=6, 60%) in trans. Six other heterozygous cases harboured the p.Asn1868Ile allele in cis with a known mutation; the remainder were in trans as determined by phase analysis in available families (table 1 and supplementary table 1). The haplotype containing the c.5603A>T (p.Asn1868Ile) allele has been established.22 In cases where phase determination was impossible due to the absence of family members, the haplotype was identified by subtracting the genotype of the known allele. Further examination established that the mutations in trans with the p.Asn1868Ile variant are deleterious, predominantly null mutations based on in silico predictions and experimental data (tables 1 and 2). The breakdown of mutations in trans with p.Asn1868Ile include the following: 6 c.5461–10T>C, 9 stop mutations, 7 small indels resulting in a frameshift, 6 severe splicing-altering variants (eg, at +/−1 and+/−2 positions) and 33 missense mutations, including well-known severe variants such as two cases of c.2894A>G (p.Asn965Ser)24 and eight cases of the complex allele c.[1622T>C;3113C>T] (p.[Leu541Pro;Ala1038Val]).25–27 Among others, seven cases with mutations of arginine to tryptophan—c.52C>T (p.Arg18Trp), c.868C>T (p.Arg290Trp), c.1804C>T (p.Arg602Trp), c.4918C>T (p.Arg1640Trp) and c.6229C>T (p.Arg2077Trp) especially stood out (table 1).
Type and frequency of the allele in trans for selected ABCA4 disease alleles
Sequencing of the entire ABCA4 genomic locus had been performed for 93/117 monoallelic ABCA4 cases as previously described.8 In many cases with a single pathogenic ABCA4 variant, another confirmed or highly likely pathogenic ABCA4 deep intronic variant(s) were identified.8 However, none of the 61 cases with the p.Asn1868Ile allele harboured any definitely or very likely pathogenic deep intronic variants, suggesting that the disease phenotype was indeed conferred by the p.Asn1868Ile variant in trans with a deleterious ABCA4 mutation.
The frequency of another presumed ‘mild’ p.Gly863Ala variant is high in populations of European descent: ~2% in populations of Dutch and Swedish ancestry22 28 and ~0.8% in all Europeans (gnomAD), therefore, it was considered hypomorphic, that is, requiring to be paired, in trans, with a ‘severe’ ABCA4 allele in order to present phenotypically.28 This is in part true, the analysis of our dataset of 643 patients (table 2) suggests that the ABCA4 allele in trans from p.Gly863Ala is deleterious or very likely deleterious in >70% of cases. However, we have encountered at least two exceptions of patients homozygous for p.Gly863Ala and in some cases the opposing allele is known to be mild, for example, the missense alleles c.5882G>A (p.Gly1961Glu), c.2947A>G (p.Thr983Ala) and c.4919G>A (Arg1640Gln) (see online supplementary table 1).
The p.Gly863Ala allele is always in cis with the p.Asn1868Ile in patients with ABCA4 disease, which lead us to initially consider the latter to be a benign allele on the same haplotype, as also observed by Maugeri et al in 2002,22 where disease-associated p.Gly863Ala haplotypes were defined. When analysing data from our large cohorts of patients with age-related macular degeneration (AMD) and matched controls,29–31 we observed that ~90% of cases with the p.Gly863Ala variant did not carry the p.Asn1868Ile allele. Analysis of these and previously published data22 revealed that the p.Asn1868Ile variant is allelic with the p.Gly863Ala in only ~13% of cases in populations of European ancestry.
Further evidence of the complex allele c.[2588G>C;5603A>T] (p.[Gly863Ala;Asn1868Ile]) being disease-associated and the p.Gly863Ala alone not, comes from the calculation of expected and observed allele frequencies in our patient cohort and matched general population. If we consider the frequency of the complex allele, p.[Gly863Ala;Asn1868Ile] to be ~13% of all cases carrying the p.Gly863Ala allele, which frequency is ~1% on average in the general population of European descent, then our cohort of 643 cases should harbour 13 cases with the p.Gly863Ala allele and 2 cases with the complex allele p.[Gly863Ala;Asn1868Ile], if neither of these would be disease-causing. In fact, we observe 35 cases (17X more of the expected at random) of the complex allele (p<0.0001) and only 4 cases (3X less of the expected at random) of the simplex allele (p<0.0001). These data additionally support the pathogenicity of the complex p.[Gly863Ala;Asn1868Ile] allele and not of the p.Gly863Ala alone.
Finally, we determined that 2 out of 643 patients were homozygous for the p.[Gly863Ala;Asn1868Ile] complex allele and none for the p.Gly863Ala allele alone. Since the population frequency of the p.Gly863Ala allele is 1% and the complex allele ~0.1%, the random occurrence of homozygotes is 1:10 000 and 1:1 000 000, respectively. Since we did not detect, among our cohort of patients with ABCA4 disease, any homozygotes for the simplex allele and found two for the complex allele, the odds for p.Gly863Ala being disease-causing without the p.Asn1868Ile allele are extremely low.
Genotype-phenotype correlations
Although previously described as clinically ‘mild’ alleles, patients harbouring the c.5882G>A (p.Gly1961Glu) or c.2588G>C (p.Gly863Ala) alleles have substantially different phenotypes. Patients with p.Gly1961Glu indeed exhibit milder disease expression,32 33 although not in the overall rate of disease progression but in a distinct phenotypic pattern that, interestingly, overlaps with patients harbouring p.Asn1868Ile. ABCA4 disease invariably begins as a maculopathy with an enlarging lesion of outer retinal atrophy and accumulation of yellow foci, or flecks, at the level of the retinal pigment epithelium (RPE). Patients with most other ABCA4 disease alleles exhibit progressively severe fleck patterns from very few in the macula to a stage of ‘absolute confluence’ across the posterior pole between the ages 30 and 40 years (figure 1).

Summary of disease trajectories associated with autofluorescent fleck accumulation in ABCA4 disease. (A) Autofluorescence images illustrating milestones of fleck accumulation beginning with confined or bull’s eye lesions with no visible flecks, to early accumulations within and around the macula, state of absolute confluence with fleck atrophy and end-stage of multiple coalescing lesions across the posterior pole. (B) Timeline of milestones (mean age) disease trajectories with respect to genotype groups. Symptomatic onset (black dashed line) varies across genotypic groups. Notably, patients harbouring c.5882G>A (p.Gly1961Glu) and c.5603A>T (p.Asn1868Ile) present at a significantly later age as compared with patients harbouring other ABCA4 variant combinations. Both c.5882G>A (p.Gly1961Glu) and c.5603A>T (p.Asn1868Ile) patient groups exhibit a delayed onset of visible flecks in the retina (blue line) and, notably, do not progress to the absolute confluence (red line) or end-stage stage (black line) phenotypes as seen in all other cases.
Patients harbouring p.Gly1961Glu or p.Asn1868Ile consistently exhibit milder spatiotemporal fleck patterns even at advanced age, which never progresses to the absolute confluence stage as illustrated by fundus autofluorescence imaging which detects lipofuscin accumulation (figure 1). The resistance to fleck accumulation as seen in p.Gly1961Glu or p.Asn1868Ile patients is further exemplified by the occurrence of a particular phenotype that is seen only in this patient group where a well-defined, unifocal lesion of dark atrophy ranging from 8.6 to 56.4 mm2 in size with proximally bordering, lesion-centric flecks appears (see online supplementary figure 1). Similar lesions of dark atrophy are typically numerous (multifocal) and observed in the background of more advanced fleck stages with most other ABCA4 disease cases (figure 1). Additionally, the individual morphology of flecks in p.Gly1961Glu or p.Asn1868Ile appear to be distinct in that they are predominantly larger in size and are well-defined in shape and generally more sparsely distributed (see online supplementary figure 1).
Despite sharing a similar phenotypic trajectory with patients carrying p.Gly1961Glu with respect to fleck formation and lesion progression, patients harbouring the p.Asn1868Ile variant also exhibit distinct clinical characteristics. ABCA4 disease is predominantly juvenile-onset as patients (including those with p.Gly1961Glu) report visual symptoms within the first and second decades of life; however, p.Asn1868Ile patients report a significantly delayed onset within the fourth decade of life (mean age=36.3 years; p<0.0001) (figure 2A). The prevalence of foveal sparing, defined as the structural and function preservation of outer retinal layers in the fovea despite the progressing atrophy of the macula, was observed in approximately one-third of patients with ABCA4 disease, but in 84.7% of p.Asn1868Ile patients (figure 2B). Visual function and acuity is often preserved in cases where the outer retinal layers of the fovea (RPE, ellipsoid zone and external limiting membrane) (figure 2C and D) are structurally intact within a region of macular atrophy, which may be connected to the delay in perception of visual symptoms within this group.

Summary of phenotypic distinction in patients harbouring the c.5603A>T (p.Asn1868Ile) allele of ABCA4. (A) Patients harbouring biallelic ABCA4 variants, including null alleles, and c.5882G>A (p.Gly1961Glu) report visual symptoms at a mean age of 19.7 and 22.7 years, respectively, while the disease is significantly delayed (to 36.3 years, p<0.0001) in patients with the c.5603A>T (p.Asn1868Ile) allele. (B) The prevalence of foveal sparing is highest among patients with p.Asn1868Ile at 84.7% while observed only in ~33% of cases with other ABCA4 variants including p.Gly1961Glu. (C) Autofluorescence imaging across the macula in a p.Asn1868Ile patient exhibiting foveal sparing within an area of retinal pigment epithelium (RPE) and photoreceptor cell atrophy. (D) A spectral domain-optical coherence tomographic scan across the fovea of the same patient reveals the presence of outer retinal layers: RPE, ellipsoid zone (EZ) and external limiting membrane (ELM) in the fovea. SD-OCT, spectral domain-optical coherence tomography.
{kind=link}
![[Supplemental_Figure_1.jpg]](https://jmg.bmj.com/content/jmedgenet/54/6/404/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
{kind=link}
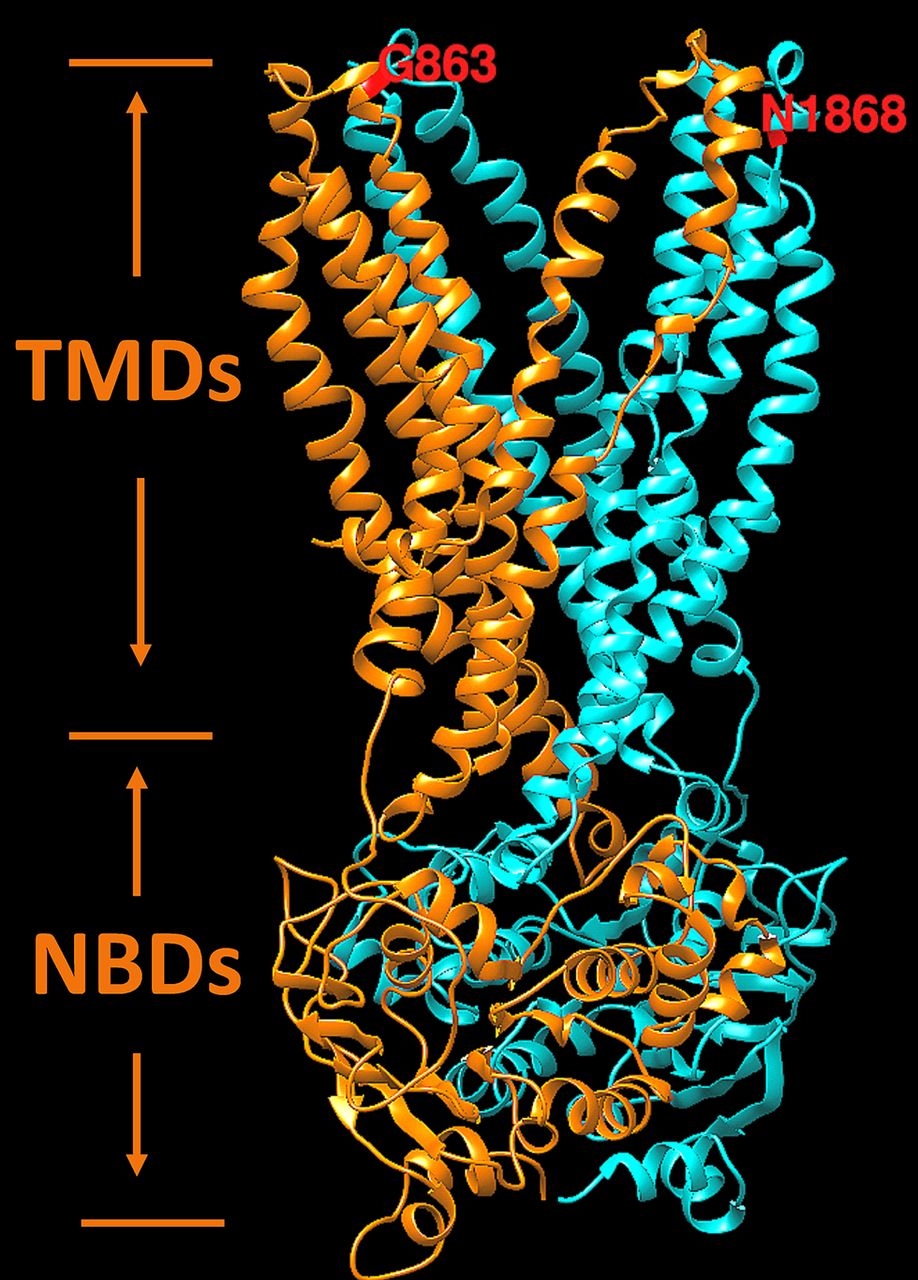
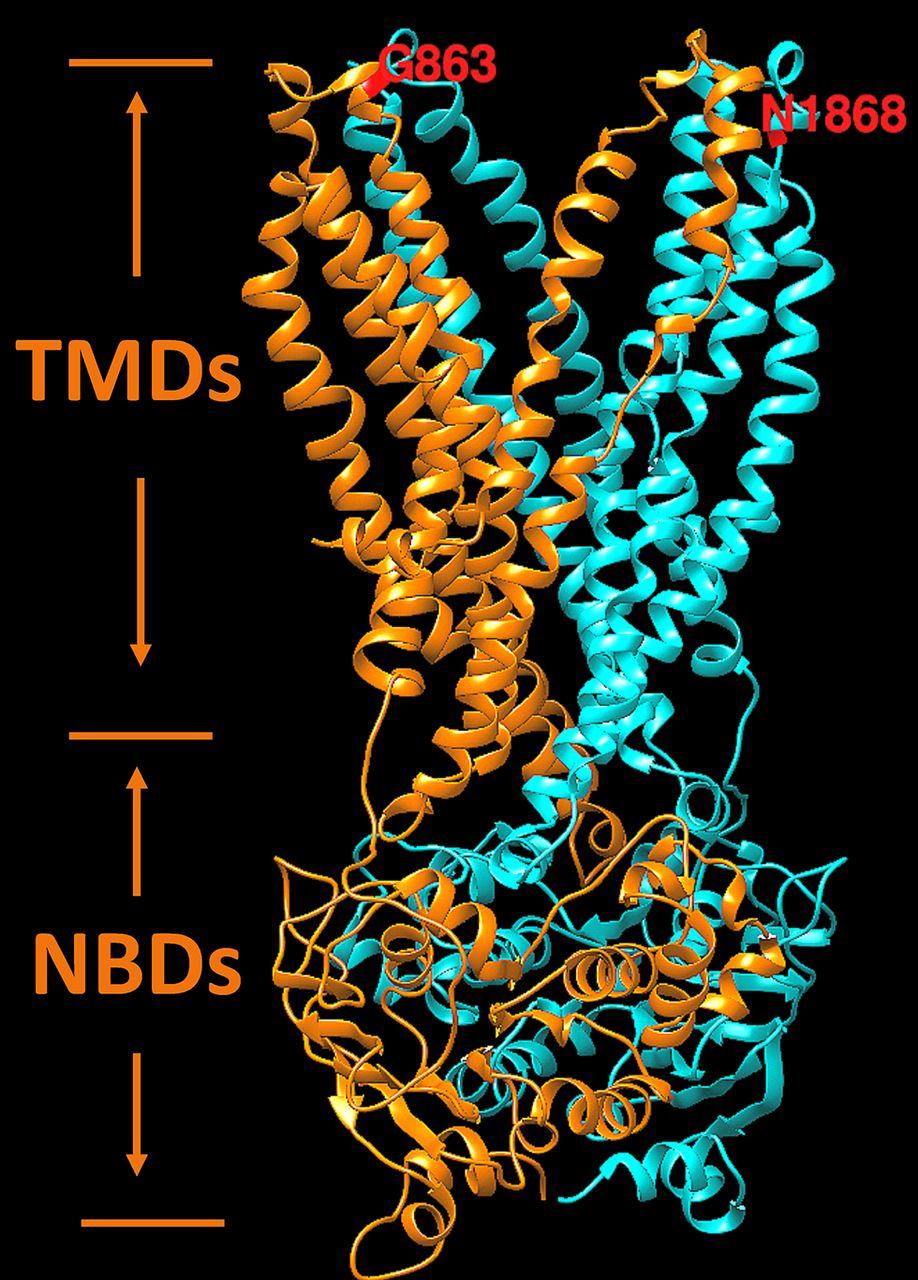
Hypothetical structural model of ABCA4 transmembrane and nucleotide-binding domains. Two symmetrical structural motifs are shown by different colours. Ribbon structure of motif 1, which include transmembrane and nucleotide-binding domains localised in the first half of the protein, is shown in orange. Motif 2 is localised in the second half of the ABCA4 protein and shown in cyan. Transmembrane and nucleotide-binding domains are labelled according to Dawson and Locher49 as the TMDs and NBDs, respectively. Residues G863 and N1868 are exposed to disk lumen. Energetic (ΔΔG, kcal/mol) and protein folding effects of structural perturbations p.Asn1868Ile and p.Gly863Ala were predicted from the model using the unfolding mutation screen.20 21
As expected, the phenotypes of biallelic patients with p.Asn1868Ile in cis with other mutations (eg, c.5461–10T>C, p.Cys1490Tyr, etc) were typically consistent with the effect of overall genotype and presented with mostly early onset, severe phenotypes. As a complex allele, p.Asn1868Ile/p.Gly863Ala acted as a fully penetrant allele with a moderate effect and resulted in variable phenotypes, which appeared to be, as expected, reflective also of the variant on the opposite allele. Intermediate phenotypes were associated with missense variants, for example, c.2947A>G (p.Thr983Ala) and c.1964T>G (p.Phe655Cys), while advanced cases were associated with deletions and splice site variants, for example, c.5778_5779del (p.Arg1927fs) and c.6088C>T (p.Arg2030*). The only two patients homozygous for the complex p.[Gly863Ala;Asn1868Ile] allele presented with later onset and milder disease and foveal sparing in one of the two cases.
Late-onset ABCA4 disease and AMD
There is an ongoing discussion about the prevalence and role of heterozygous ABCA4 alleles in a late-onset, common disease, AMD.25 31 34–37 While the answer is still not unequivocal—some studies find statistically significantly elevated ABCA4 alleles in AMD and some do not—it has been suggested that many of AMD cases are actually misdiagnosed late-onset patients with ABCA4 disease.37 38 It has also been suggested that patients with late-onset ABCA4 disease remain monoallelic after complete ABCA4 sequencing much more often than those with early onset disease.38 Finally, data on phenotypes of individuals who represent true carriers, such as parents of patients with ABCA4 disease, are also very conflicting, where some, including us39 suggest that there is no detectable disease phenotype or increased lipofuscin accumulation, at least up to 60 years of age, some other studies have frequently found disease phenotypes in parents of STGD1 patients.40 41 While unequivocal answers to these questions are beyond of the scope of this study, we queried the frequencies and composition of ABCA4 alleles in late-onset ABCA4 disease, in patients with AMD, and in matched controls.
We defined late onset as ≥45 years of age, in accordance with previous studies,38 who had found in a small cohort of 21 unrelated patients that a greater proportion of late-onset cases were monoallelic (52%) than biallelic (38%) from which the authors proposed that slower disease progression from defects on just one disease allele may eventually cause ABCA4 disease. When examining our larger group of patients for whom similar disease onset data were available, an even larger disparity among monoallelic and biallelic late-onset cases; 27.6% vs 7.7%, respectively, was observed. However, following the discovery of p.Asn1868Ile to be the second causal variant in ~80% of these monoallelic late-onset cases, a similar fraction of the late-onset phenotype (~10%) was observed in both monoallelic and biallelic cases of the late-onset disease. The remaining missing alleles in late onset, monoallelic disease cases could be another hypomorphic variant or, more likely, another yet unknown modifier allele. Our data therefore do not corroborate an association between late-onset ABCA4 disease phenotype and monoallelic cases. Genetic analysis taking into account the p.Asn1868Ile variant would allow distinguishing true monoallelic carriers affected with AMD from biallelic late-onset ABCA4 disease in most cases under investigation.
Discussion
Since the ABCA4 gene was cloned 20 years ago,1 the ABCA4 locus continues to be the most heterogeneous of Mendelian eye diseases in terms of both genetic and phenotypic expression.42 While loci with similar genetic heterogeneity are not rare—the related ABC transporter, CFTR, locus presents with more pathogenic variants,43 the ABCA4 gene continues to emerge as an example of complexity in Mendelian disorders. Due to a high prevalence of disease-associated alleles, the variation in the ABCA4 locus underlies a wide spectrum of disease phenotypes, from early onset, rapidly progressing disease resulting in panretinal degeneration to late-onset, milder disease with limited visual deterioration that can phenotypically overlap with the more prevalent AMD. The c.5603A>T (p.Asn1868Ile) allele is, pending unequivocal functional evidence, one of the most frequent alleles (MAF >6.5% in the matched general population) in Mendelian diseases which causes a phenotype. The ABCA4 locus has several frequent disease-causing alleles in some populations. The best known is the p.Gly1961Glu variant, with the frequency as high as 10% in Somalian population.44 While several compound heterozygous and even homozygous cases of ABCA4 disease due to the p.Gly1961Glu allele of Somalian ancestry have been described,32 the penetrance of the variant is unknown in that population since very few cases of Somalian ancestry have been screened due to limited access to patients. However, it is a fully penetrant hypomorphic allele in all other populations where significant numbers of patients have been sequenced. For example, the frequency of the p.Gly1961Glu in South Asian (Indian, etc) populations is 1.5% and we detected it in ~50% of patients (~24% of alleles) with ABCA4 disease from India.45 Another recently described frequent variant is the c.6320G>A (p.Arg2107His) allele, with MAF ~2% in African-Americans and MAF >19% in patients of AA descent.46
Interestingly, while this study unequivocally demonstrates the non-pathogenicity of the p.Gly863Ala variant alone, that is, without p.Asn1868Ile, it was shown to have an effect in in vitro assays where less protein production, reduced ATP binding and hydrolysis47 and reduced retinal transfer48 were demonstrated. It has also been shown to have a dual effect: it either results in a deletion of Gly863, due to aberrant splicing, or a substitution of the same glycine to alanine.28 The p.Asn1868Ile variant, on the contrary, has been showing minor, if any effects in the ATP binding and hydrolysis assays, which were comparable to the wild-type protein.47 Many reasons could explain these discrepancies, the foremost being the in vitro assays likely not adequately recapitulating the in vivo conditions. One of known issues with the in vitro protein assays are the possible detrimental effects of detergent solubilisation on the stability of certain ABCA4 variants. Functional studies of knock-in mouse models, which will bypass the issues with in vitro assays, are under way to assess the effects of the p.Asn1868Ile, p.Gly863Ala, and the complex allele carrying both variants. Here, the influence of the variants on protein structure and function were assessed by prediction from the atomic level of protein structure as previously described for X linked retinoschisin.18 19 The hypothetical structure of transmembrane and nucleotide-binding domains of the ABCA4 protein responsible for the flipping of N-retinylidene-PE across photoreceptor disc membranes is shown in figure 3. In this model, both variants, p.Asn1868Ile and p.Gly863Ala, are located on the same side the of the photoreceptor outer segment disc membrane and both were predicted to affect the protein folding by the unfolding mutation screen.20 21 While not directly experimentally demonstrated, it is predicted and very plausible that both variants forming the complex allele will have a synergistic effect (figure 3).
In fact, the severity of many missense mutations, in addition to those already proven by functional assays or statistical analyses in large patient cohorts (p.[Leu541Pro;Ala1038Val] p.Cys1490Tyr, p.Asn965Ser, p.Cys54Tyr, etc)24 26 27 47 can be assessed by their presence in trans with the p.Asn1868Ile variant, which acts as a ‘litmus test’ for severity of an ABCA4 variant. By applying this test and in silico analysis, missense mutations determined to be deleterious included c.3261A>C (p.Glu1087Asp), c.3259G>A (p.Glu1087Lys), c.5395A>G (p.Asn1799Asp), c.1921T>C (p.Cys641Arg), c.262G>A (p.Gly88Arg) and c.5923G>C (p.Gly1975Arg).
Conclusions
First, our study identified the conditions under which the very frequent, previously considered benign c.5603A>T (p.Asn1868Ile) variant is pathogenic. It is an ‘extremely’ hypomorphic allele and is phenotypically expressed only when in trans with a deleterious mutation.
Second, the third most frequent ABCA4 mutation, the c.2588G>C (p.Gly863Ala) variant, is not disease causing alone, but rather requires the c.5603A>T (p.Asn1868Ile) variant in cis for pathogenicity. Therefore, the p.Gly863Ala variant acts not as a disease-associated hypomorphic variant, but rather as a modifier, since it causes disease only when in cis with p.Asn1868Ile.
Third, the phenotypes caused by the p.Asn1868Ile variant are late onset, frequently associated with foveal sparing and distinguishable from other ABCA4 disease phenotypes.
Fourth, the cases compound heterozygous for p.Asn1868Ile account for at least 10% of all known ABCA4 disease and refine another 10% where they form a complex allele with the p.Gly863Ala variant.
Fifth, the p.Asn1868Ile alleles account for >50% of previously considered ‘one mutation’ cases of ABCA4 disease and for a very large fraction (~80%) of late-onset cases. While it had been suggested before that many late-onset cases are monoallelic, these are in fact biallelic and harbour the extremely hypomorphic p.Asn1868Ile variant. Therefore, the p.Asn1868Ile variant explains most of the late-onset biallelic ABCA4 disease sometimes masquerading as AMD.
In summary, these data suggest a substantial revision of genetic screening and assessment of pathogenic variants in ABCA4 disease including the absolute necessity of determining of the phase of all alleles. It also serves as an example for all other Mendelian diseases, where careful consideration should be given to seemingly benign and (very) frequent variants, and to specific combinations of these.
References
Footnotes
Contributors All the authors contributed significantly to this research. Study conceptualisation and design: RA. Sequencing: JZ. Genetic analysis and interpretation: RA and JZ. Acquisition of clinical data and specimens: WL and FTC. Clinical analysis and interpretation: WL, FTC, SHT, GF and KS. Drafting of manuscript: RA, JZ and WL. Critical revisions: All authors. All authors agree to be accountable for all aspects of the work.
Funding This work was supported, in part, by grants from the National Eye Institute/NIH EY021163, EY019861 and EY019007 (Core Support for Vision Research); Pangere Family Foundation, Pangere Center, Chicago Lighthouse, OPOS Stiftung zugunsten Wahrnehmungsbehinderten, St. Gallen, Switzerland and Alfred-Vogt-Stifung, St. Gallen, Switzerland and unrestricted funds from Research to Prevent Blindness (New York, NY) to the Department of Ophthalmology, Columbia University.
Competing interests None declared.
Patient consent All data used and presented in the study cannot be traced or identified to an individual subject. All identifiable information were removed in accordance with the Health Insurance Portability and Accountability Act of 1996 regulations and Columbia University Medical Center Institutional Review Board protocols.
Ethics approval Columbia University Medical Center Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.